1.12: An Introduction to Energy Dispersive X-ray Spectroscopy
- Page ID
- 55830

Introduction
Energy-dispersive X-ray spectroscopy (EDX or EDS) is an analytical technique used to probe the composition of a solid materials. Several variants exist, but the all rely on exciting electrons near the nucleus, causing more distant electrons to drop energy levels to fill the resulting “holes.” Each element emits a different set of X-ray frequencies as their vacated lower energy states are refilled, so measuring these emissions can provide both qualitative and quantitative information about the near-surface makeup of the sample. However, accurate interpretation of this data is dependent on the presence of high-quality standards, and technical limitations can compromise the resolution.
Physical Underpinnings
In the quantum mechanical model of the atom, an electron’s energy state is defined by a set of quantum numbers. The primary quantum number, n, provides the coarsest description of the electron’s energy level, and all the sublevels that share the same primary quantum number are sometimes said to comprise an energy “shell.” Instead of describing the lowest-energy shell as the “n = 1 shell,” it is more common in spectroscopy to use alphabetical labels: The K shell has n = 1, the L shell has n = 2, the M shell has n = 3, and so on. Subsequent quantum numbers divide the shells into subshells: one for K, three for L, and five for M. Increasing primary quantum numbers correspond with increasing average distance from the nucleus and increasing energy (Figure \(\PageIndex{1}\)). An atom’s core shells are those with lower primary quantum numbers than the highest occupied shell, or valence shell.

Transitions between energy levels follow the law of conservation of energy. Excitation of an electron to a higher energy state requires an input of energy from the surroundings, and relaxation to a lower energy state releases energy to the surroundings. One of the most common and useful ways energy can be transferred into and out of an atom is by electromagnetic radiation. Core shell transitions correspond to radiation in the X-ray portion of the spectrum; however, because the core shells are normally full by definition, these transitions are not usually observed.
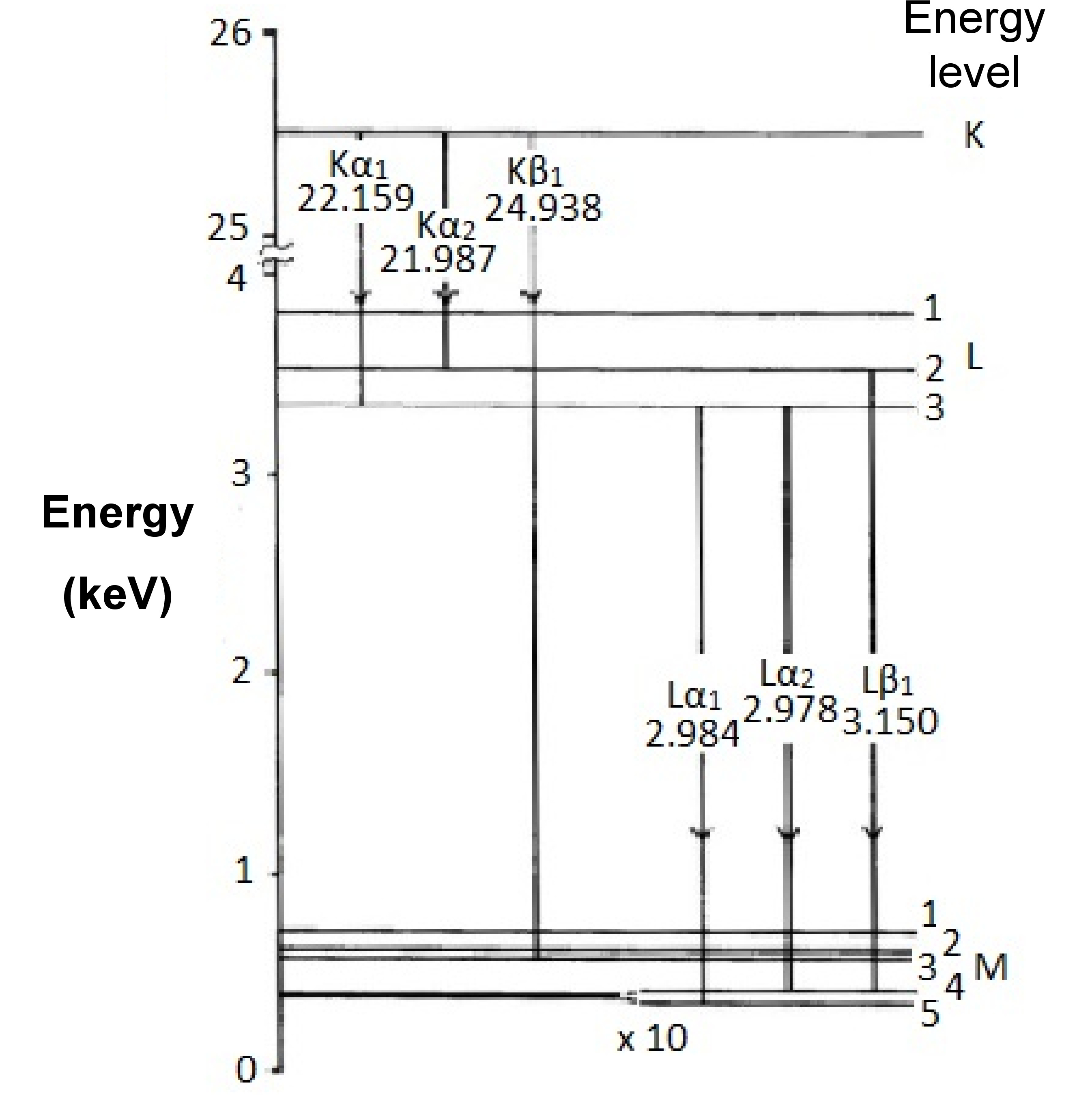
X-ray spectroscopy uses a beam of electrons or high-energy radiation (see instrument variations, below) to excite core electrons to high energy states, creating a low-energy vacancy in the atoms’ electronic structures. This leads to a cascade of electrons from higher energy levels until the atom regains a minimum-energy state. Due to conservation of energy, the electrons emit X-rays as they transition to lower energy states. It is these X-rays that are being measured in X-ray spectroscopy. The energy transitions are named using the letter of the shell where ionization first occurred, a Greek letter denoting the group of lines that transition belongs to, in order of decreasing importance, and a numeric subscript ranking the peak's the intensity within that group. Thus, the most intense peak resulting from ionization in the K shell would be Kα1 (Figure \(\PageIndex{2}\)). Since each element has a different nuclear charge, the energies of the core shells and, more importantly, the spacing between them vary from one element to the next. While not every peak in an element’s spectrum is exclusive to that element, there are enough characteristic peaks to be able to determine composition of the sample, given sufficient resolving power.
Instrumentation and Sample Preparation
Instrument variations
There are two common methods for exciting the core electrons off the surface atoms. The first is to use a high-energy electron beam like the one in a scanning electron microscope (SEM). The beam is produced by an electron gun, in which electrons emitted thermionically from a hot cathode are guided down the column by an electric field and focused by a series of negatively charged “lenses.” X-rays emitted by the sample strike a lithium-drifted silicon p-i-n junction plate. This promotes electrons in the plate into the conduction band, inducing a voltage proportional to the energy of the impacting X-ray which generally falls between about 1 and 10 keV. The detector is cooled to liquid nitrogen temperatures to reduce electronic noise from thermal excitations.
It is also possible to use X-rays to excite the core electrons to the point of ionization. In this variation, known as energy-dispersive X-ray fluorescence analysis (EDXRFA or XRF), the electron column is replaced by an X-ray tube and the X-rays emitted by the sample in response to the bombardment are called secondary X-rays, but these variants are otherwise identical.
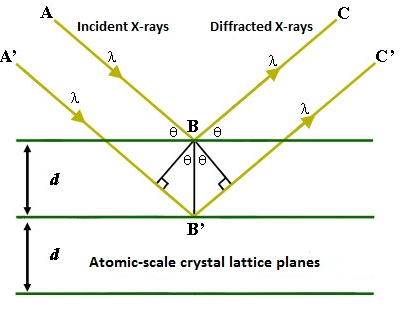
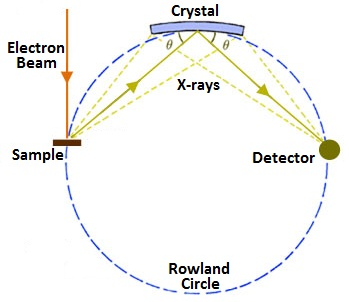
Regardless of the excitation method, subsequent interactions between the emitted X-rays and the sample can lead to poor resolution in the X-ray spectrum, producing a Gaussian-like curve instead of a sharp peak. Indeed, this spreading of energy within the sample combined with the penetration of the electron or X-ray beam leads to the analysis of a roughly 1 µm3 volume instead of only the surface features. Peak broadening can lead to overlapping peaks and a generally misleading spectrum. In cases where a normal EDS spectrum is inadequately resolved, a technique called wavelength-dispersive X-ray spectroscopy (WDS) can be used. The required instrument is very similar to the ones discussed above, and can use either excitation method. The major difference is that instead of having the X-rays emitted by the sample hit the detector directly, they first encounter an analytical crystal of know lattice dimensions. Bragg’s law predicts that the strongest reflections off the crystal will occur for wavelengths such that the path difference between a rays reflecting from consecutive layers in the lattice is equal to an integral number of wavelengths. This is represented mathematically as \ref{1}, where n is an integer, λ is the wavelength of impinging light, d is the distance between layers in the lattice, and θ is the angle of incidence. The relevant variables for the equation are labeled in Figure \(\PageIndex{3}\).
\[ n\lambda \ =\ 2d\ sin\ \theta \label{1} \]
By moving the crystal and the detector around the Rowland circle, the spectrometer can be tuned to examine specific wavelengths (\ref{1}). Generally, an initial scan across all wavelengths is taken first, and then the instrument is programmed to more closely examine the wavelengths that produced strong peaks. The resolution available with WDS is about an order of magnitude better than with EDS because the analytical crystal helps filter out the noise of subsequent, non-characteristic interactions. For clarity, “X-ray spectroscopy” will be used to refer to all of the technical variants just discussed, and points made about EDS will hold true for XRF unless otherwise noted.
Sample Preparation
Compared with some analytical techniques, the sample preparation required for X-ray spectroscopy or any of the related methods just discussed is trivial. The sample must be stable under vacuum, since the sample chamber is evacuated to prevent the atmosphere from interfering with the electron beam or X-rays. It is also advisable to have the surface as clean as possible; X-ray spectroscopy is a near-surface technique, so it should analyze the desired material for the most part regardless, but any grime on the surface will throw off the composition calculations. Simple qualitative readings can be obtained from a solid of any thickness, as long as it fits in the machine, but for reliable quantitative measurements, the sample should be shaved as thin as possible.
Data Interpretation
Qualitative analysis, the determination of which elements are present in the sample but not necessarily the stoichiometry, relies on empirical standards. The energies of the commonly used core shell transitions have been tabulated for all the natural elements. Since combinations of elements can act differently than a single element alone, standards with compositions as similar as possible to the suspected makeup of the sample are also employed. To determine the sample’s composition, the peaks in the spectrum are matched with peaks from the literature or standards.
Quantitative analysis, the determination of the sample’s stoichiometry, needs high resolution to be good enough that the ratio of the number of counts at each characteristic frequency gives the ratio of those elements in the sample. It takes about 40,000 counts for the spectrum to attain a 2σ precision of ±1%. It is important to note, however, that this is not necessarily the same as the empirical formula, since not all elements are visible. Spectrometers with a beryllium window between the sample and the detector typically cannot detect anything lighter than sodium. Spectrometers equipped with polymer based windows can quantify elements heavier than beryllium. Either way, hydrogen cannot be observed by X-ray spectroscopy.
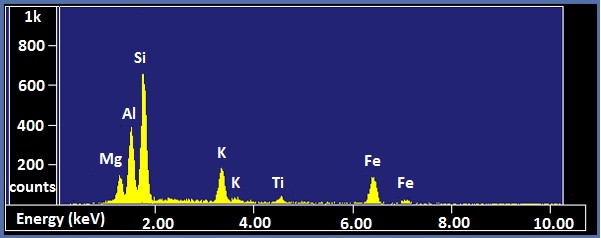
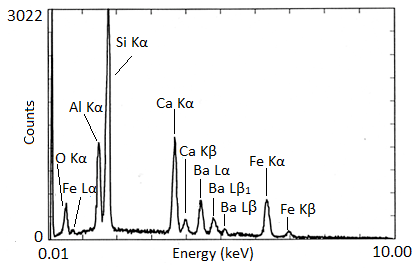
X-ray spectra are presented with energy in keV on the x-axis and the number of counts on the y-axis. The EDX spectra of biotite and NIST glass K309 are shown as examples (Figure \(\PageIndex{5}\) and Figure \(\PageIndex{6}\) respectively). Biotite is a mineral similar to mica which has the approximate chemical formula K(Mg,Fe)3AlSi3O10(F,OH)2. Strong peaks for manganese, aluminum, silicon, potassium, and iron can be seen in the spectrum. The lack of visible hydrogen is expected, and the absence of oxygen and fluorine peaks suggests the instrument had a beryllium window. The titanium peak is small and unexpected, so it may only be present in trace amounts. K309 is a mix of glass developed by the National Institute for Standards and Technology. The spectrum shows that it contains significant amounts of silicon, aluminum, calcium, oxygen, iron, and barium. The large peak at the far left is the carbon signal from the carbon substrate the glass was placed on.
Limitations
As has just been discussed, X-ray spectroscopy is incapable of seeing elements lighter than boron. This is a problem given the abundance of hydrogen in natural and man-made materials. The related techniques X-ray photoelectron spectroscopy (XPS) and Auger spectroscopy are able to detect Li and Be, but are likewise unable to measure hydrogen.
X-ray spectroscopy relies heavily on standards for peak identification. Because a combination of elements can have noticeably different properties from the individual constituent elements in terms of X-ray fluorescence or absorption, it is important to use a standard as compositionally similar to the sample as possible. Naturally, this is more difficult to accomplish when examining new materials, and there is always a risk of the structure of the sample being appreciably different than expected.
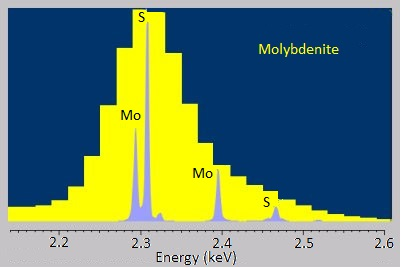
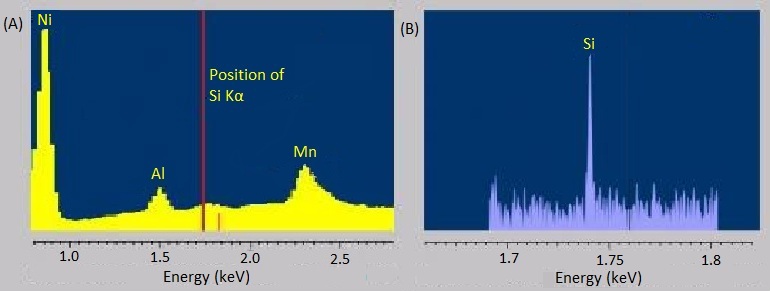
The energy-dispersive variants of X-ray spectroscopy sometimes have a hard time distinguishing between emissions that are very near each other in energy or distinguishing peaks from trace elements from background noise. Fortunately, the wavelength-dispersive variants are much better at both of these. The rough, stepwise curve in Figure \(\PageIndex{7}\) represents the EDS spectrum of molybdenite, a mineral with the chemical formula MoS2. Broadened peaks make it difficult to distinguish the molybdenum signals from the sulfur ones. Because WDS can select specific wavelengths, it has much better resolution and can pinpoint the separate peaks more accurately. Similarly, the trace silicon signal in the EDS spectrum of the nickel-aluminum-manganese alloy in Figure \(\PageIndex{8}\)a is barely distinguishable as a bump in the baseline, but the WDS spectrum in Figure \(\PageIndex{8}\)b clearly picks it up.