
1.2: Beer’s Law
- Page ID
- 111324

What factors influence the absorbance that you would measure for a sample? Is each factor directly or inversely proportional to the absorbance?
One factor that influences the absorbance of a sample is the concentration (c). The expectation would be that, as the concentration goes up, more radiation is absorbed and the absorbance goes up. Therefore, the absorbance is directly proportional to the concentration.
A second factor is the path length (b). The longer the path length, the more molecules there are in the path of the beam of radiation, therefore the absorbance goes up. Therefore, the path length is directly proportional to the concentration.
When the concentration is reported in moles/liter and the path length is reported in centimeters, the third factor is known as the molar absorptivity (\(\varepsilon\)). In some fields of work, it is more common to refer to this as the extinction coefficient. When we use a spectroscopic method to measure the concentration of a sample, we select out a specific wavelength of radiation to shine on the sample. As you likely know from other experiences, a particular chemical species absorbs some wavelengths of radiation and not others. The molar absorptivity is a measure of how well the species absorbs the particular wavelength of radiation that is being shined on it. The process of absorbance of electromagnetic radiation involves the excitation of a species from the ground state to a higher energy excited state. This process is described as an excitation transition, and excitation transitions have probabilities of occurrences. It is appropriate to talk about the degree to which possible energy transitions within a chemical species are allowed. Some transitions are more allowed, or more favorable, than others. Transitions that are highly favorable or highly allowed have high molar absorptivities. Transitions that are only slightly favorable or slightly allowed have low molar absorptivities. The higher the molar absorptivity, the higher the absorbance. Therefore, the molar absorptivity is directly proportional to the absorbance.
If we return to the experiment in which a spectrum (recording the absorbance as a function of wavelength) is recorded for a compound for the purpose of identification, the concentration and path length are constant at every wavelength of the spectrum. The only difference is the molar absorptivities at the different wavelengths, so a spectrum represents a plot of the relative molar absorptivity of a species as a function of wavelength.
Since the concentration, path length and molar absorptivity are all directly proportional to the absorbance, we can write the following equation, which is known as the Beer-Lambert law (often referred to as Beer’s Law), to show this relationship.
\[\mathrm{A = \varepsilon bc} \nonumber \]
Note that Beer’s Law is the equation for a straight line with a y-intercept of zero.
If you wanted to measure the concentration of a particular species in a sample, describe the procedure you would use to do so.
Measuring the concentration of a species in a sample involves a multistep process.
One important consideration is the wavelength of radiation to use for the measurement. Remember that the higher the molar absorptivity, the higher the absorbance. What this also means is that the higher the molar absorptivity, the lower the concentration of species that still gives a measurable absorbance value. Therefore, the wavelength that has the highest molar absorptivity (\(\lambda\)max) is usually selected for the analysis because it will provide the lowest detection limits. If the species you are measuring is one that has been commonly studied, literature reports or standard analysis methods will provide the \(\lambda\)max value. If it is a new species with an unknown \(\lambda\)max value, then it is easily measured by recording the spectrum of the species. The wavelength that has the highest absorbance in the spectrum is \(\lambda\)max.
The second step of the process is to generate a standard curve. The standard curve is generated by preparing a series of solutions (usually 3-5) with known concentrations of the species being measured. Every standard curve is generated using a blank. The blank is some appropriate solution that is assumed to have an absorbance value of zero. It is used to zero the spectrophotometer before measuring the absorbance of the standard and unknown solutions. The absorbance of each standard sample at \(\lambda\)max is measured and plotted as a function of concentration. The plot of the data should be linear and should go through the origin as shown in the standard curve in Figure \(\PageIndex{2}\). If the plot is not linear or if the y-intercept deviates substantially from the origin, it indicates that the standards were improperly prepared, the samples deviate in some way from Beer’s Law, or that there is an unknown interference in the sample that is complicating the measurements. Assuming a linear standard curve is obtained, the equation that provides the best linear fit to the data is generated.

Note that the slope of the line of the standard curve in Figure \(\PageIndex{2}\) is (\(\varepsilon\)b) in the Beer’s Law equation. If the path length is known, the slope of the line can then be used to calculate the molar absorptivity.
The third step is to measure the absorbance in the sample with an unknown concentration. The absorbance of the sample is used with the equation for the standard curve to calculate the concentration.
Suppose a small amount of stray radiation (PS) always leaked into your instrument and made it to your detector. This stray radiation would add to your measurements of Po and P. Would this cause any deviations to Beer's law? Explain.
The way to think about this question is to consider the expression we wrote earlier for the absorbance.
\[\mathrm{A = \log\left(\dfrac{P_o}{P}\right)} \nonumber \]
Since stray radiation always leaks in to the detector and presumably is a fixed or constant quantity, we can rewrite the expression for the absorbance including terms for the stray radiation. It is important to recognize that Po, the power from the radiation source, is considerably larger than \(P_S\). Also, the numerator (Po + Ps) is a constant at a particular wavelength.
\[\mathrm{A = \log\left(\dfrac{P_o + P_s}{P + P_s}\right)} \nonumber \]
Now let’s examine what happens to this expression under the two extremes of low concentration and high concentration. At low concentration, not much of the radiation is absorbed and P is not that much different than Po. Since \(P_o \gg P_S\), \(P\) will also be much greater than \(P_S\). If the sample is now made a little more concentrated so that a little more of the radiation is absorbed, P is still much greater than PS. Under these conditions the amount of stray radiation is a negligible contribution to the measurements of Po and P and has a negligible effect on the linearity of Beer’s Law.
As the concentration is raised, P, the radiation reaching the detector, becomes smaller. If the concentration is made high enough, much of the incident radiation is absorbed by the sample and P becomes much smaller. If we consider the denominator (P + PS) at increasing concentrations, P gets small and PS remains constant. At its limit, the denominator approaches PS, a constant. Since Po + PS is a constant and the denominator approaches a constant (Ps), the absorbance approaches a constant. A plot of what would occur is shown in Figure \(\PageIndex{3}\).

The ideal plot is the straight line. The curvature that occurs at higher concentrations that is caused by the presence of stray radiation represents a negative deviation from Beer’s Law.
The derivation of Beer's Law assumes that the molecules absorbing radiation don't interact with each other (remember that these molecules are dissolved in a solvent). If the analyte molecules interact with each other, they can alter their ability to absorb the radiation. Where would this assumption break down? Guess what this does to Beer's law?
The sample molecules are more likely to interact with each other at higher concentrations, thus the assumption used to derive Beer’s Law breaks down at high concentrations. The effect, which we will not explain in any more detail in this document, also leads to a negative deviation from Beer’s Law at high concentration.
Beer's law also assumes purely monochromatic radiation. Describe an instrumental set up that would allow you to shine monochromatic radiation on your sample. Is it possible to get purely monochromatic radiation using your set up? Guess what this does to Beer's law.
Spectroscopic instruments typically have a device known as a monochromator. There are two key features of a monochromator. The first is a device to disperse the radiation into distinct wavelengths. You are likely familiar with the dispersion of radiation that occurs when radiation of different wavelengths is passed through a prism. The second is a slit that blocks the wavelengths that you do not want to shine on your sample and only allows \(\lambda\)max to pass through to your sample as shown in Figure \(\PageIndex{4}\).
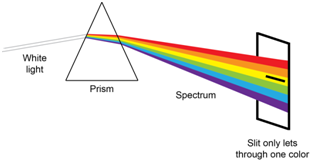
An examination of Figure \(\PageIndex{4}\) shows that the slit has to allow some “packet” of wavelengths through to the sample. The packet is centered on \(\lambda\)max, but clearly nearby wavelengths of radiation pass through the slit to the sample. The term effective bandwidth defines the packet of wavelengths and it depends on the slit width and the ability of the dispersing element to divide the wavelengths. Reducing the width of the slit reduces the packet of wavelengths that make it through to the sample, meaning that smaller slit widths lead to more monochromatic radiation and less deviation from linearity from Beer’s Law.
Is there a disadvantage to reducing the slit width?
The important thing to consider is the effect that this has on the power of radiation making it through to the sample (Po). Reducing the slit width will lead to a reduction in Po and hence P. An electronic measuring device called a detector is used to monitor the magnitude of Po and P. All electronic devices have a background noise associated with them (rather analogous to the static noise you may hear on a speaker and to the discussion of stray radiation from earlier that represents a form of noise). Po and P represent measurements of signal over the background noise. As Po and P become smaller, the background noise becomes a more significant contribution to the overall measurement. Ultimately the background noise restricts the signal that can be measured and detection limit of the spectrophotometer. Therefore, it is desirable to have a large value of Po. Since reducing the slit width reduces the value of Po, it also reduces the detection limit of the device. Selecting the appropriate slit width for a spectrophotometer is therefore a balance or tradeoff of the desire for high source power and the desire for high monochromaticity of the radiation.
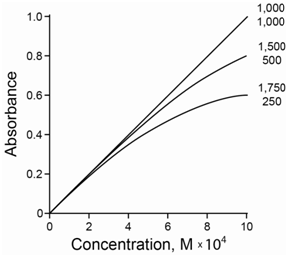
It is not possible to get purely monochromatic radiation using a dispersing element with a slit. Usually the sample has a slightly different molar absorptivity for each wavelength of radiation shining on it. The net effect is that the total absorbance added over all the different wavelengths is no longer linear with concentration. Instead a negative deviation occurs at higher concentrations due to the polychromicity of the radiation. Furthermore, the deviation is more pronounced the greater the difference in the molar absorbtivity. Figure \(\PageIndex{5}\) compares the deviation for two wavelengths of radiation with molar absorptivities that are (a) both 1,000, (b) 500 and 1,500, and (c) 250 and 1,750. As the molar absorptivities become further apart, a greater negative deviation is observed.
Therefore, it is preferable to perform the absorbance measurement in a region of the spectrum that is relatively broad and flat. The hypothetical spectrum in Figure \(\PageIndex{6}\) shows a species with two wavelengths that have the same molar absorptivity. The peak at approximately 250 nm is quite sharp whereas the one at 330 nm is rather broad. Given such a choice, the broader peak will have less deviation from the polychromaticity of the radiation and is less prone to errors caused by slight misadjustments of the monochromator.

Consider the relative error that would be observed for a sample as a function of the transmittance or absorbance. Is there a preferable region in which to measure the absorbance? What do you think about measuring absorbance values above 1?
It is important to consider the error that occurs at the two extremes (high concentration and low concentration). Our discussion above about deviations to Beer’s Law showed that several problems ensued at higher concentrations of the sample. Also, the point where only 10% of the radiation is transmitted through the sample corresponds to an absorbance value of 1. Because of the logarithmic relationship between absorbance and transmittance, the absorbance values rise rather rapidly over the last 10% of the radiation that is absorbed by the sample. A relatively small change in the transmittance can lead to a rather large change in the absorbance at high concentrations. Because of the substantial negative deviation to Beer’s law and the lack of precision in measuring absorbance values above 1, it is reasonable to assume that the error in the measurement of absorbance would be high at high concentrations.
At very low sample concentrations, we observe that Po and P are quite similar in magnitude. If we lower the concentration a bit more, P becomes even more similar to Po. The important realization is that, at low concentrations, we are measuring a small difference between two large numbers. For example, suppose we wanted to measure the weight of a captain of an oil tanker. One way to do this is to measure the combined weight of the tanker and the captain, then have the captain leave the ship and measure the weight again. The difference between these two large numbers would be the weight of the captain. If we had a scale that was accurate to many, many significant figures, then we could possibly perform the measurement in this way. But you likely realize that this is an impractical way to accurately measure the weight of the captain and most scales do not have sufficient precision for an accurate measurement. Similarly, trying to measure a small difference between two large signals of radiation is prone to error since the difference in the signals might be on the order of the inherent noise in the measurement. Therefore, the degree of error is expected to be high at low concentrations.
The discussion above suggests that it is best to measure the absorbance somewhere in the range of 0.1 to 0.8. Solutions of higher and lower concentrations have higher relative error in the measurement. Low absorbance values (high transmittance) correspond to dilute solutions. Often, other than taking steps to concentrate the sample, we are forced to measure samples that have low concentrations and must accept the increased error in the measurement. It is generally undesirable to record absorbance measurements above 1 for samples. Instead, it is better to dilute such samples and record a value that will be more precise with less relative error.
Another question that arises is whether it is acceptable to use a non-linear standard curve. As we observed earlier, standard curves of absorbance versus concentration will show a non-linearity at higher concentrations. Such a non-linear plot can usually be fit using a higher order equation and the equation may predict the shape of the curve quite accurately. Whether or not it is acceptable to use the non-linear portion of the curve depends in part on the absorbance value where the non-linearity starts to appear. If the non-linearity occurs at absorbance values higher than one, it is usually better to dilute the sample into the linear portion of the curve because the absorbance value has a high relative error. If the non-linearity occurs at absorbance values lower than one, using a non-linear higher order equation to calculate the concentration of the analyte in the unknown may be acceptable.
One thing that should never be done is to extrapolate a standard curve to higher concentrations. Since non-linearity will occur at some point, and there is no way of knowing in advance when it will occur, the absorbance of any unknown sample must be lower than the absorbance of the highest concentration standard used in the preparation of the standard curve. It is also not desirable to extrapolate a standard curve to lower concentrations. There are occasions when non-linear effects occur at low concentrations. If an unknown has an absorbance that is below that of the lowest concentration standard of the standard curve, it is preferable to prepare a lower concentration standard to ensure that the curve is linear over such a concentration region.
Another concern that always exists when using spectroscopic measurements for compound quantification or identification is the potential presence of matrix effects. The matrix is everything else that is in the sample except for the species being analyzed. A concern can occur when the matrix of the unknown sample has components in it that are not in the blank solution and standards. Components of the matrix can have several undesirable effects.
What are some examples of matrix effects and what undesirable effect could each have that would compromise the absorbance measurement for a sample with an unknown concentration?
One concern is that a component of the matrix may absorb radiation at the same wavelength as the analyte, giving a false positive signal. Particulate matter in a sample will scatter the radiation, thereby reducing the intensity of the radiation at the detector. Scattered radiation will be confused with absorbed radiation and result in a higher concentration than actually occurs in the sample.
Another concern is that some species have the ability to change the value of \(\lambda\)max. For some species, the value of \(\lambda\)max can show a pronounced dependence on pH. If this is a consideration, then all of the standard and unknown solutions must be appropriately buffered. Species that can hydrogen bond or metal ions that can form donor-acceptor complexes with the analyte may alter the position of \(\lambda\)max. Changes in the solvent can affect \(\lambda\)max as well.