
Cisplatin 12. Modes of Action of Cisplatin
- Page ID
- 2909

The discovery of cisplatin (cis-diamminedichloroplatinum, or cis-DDP) in the early 1960s generated a tremendous amount of research activity as scientists strove to understand how the drug worked in the human body to destroy cancer cells. We now believe that cisplatin coordinates to DNA and that this coordination complex not only inhibits replication and transcription of DNA, but also leads to programmed cell death (called apoptosis).
As it turns out, however, formation of any platinated coordination complex with DNA is not sufficient for cytotoxic (that is, cell-killing) activity. The corresponding trans isomer of cisplatin (namely, trans-DDP) also forms a coordination complex with DNA but unlike cisplatin, trans-DDP is not an effective chemotherapeutic agent.
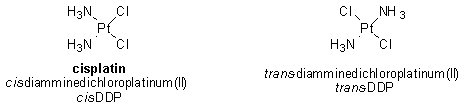
Due to the difference in geometry between cis- and trans-DDP, the types of coordination complexes formed by the two compounds with DNA are not the same. It appears that these differences are critically important in determining the efficacy of a particular compound for the treatment of cancer. For this reason, a great deal of effort has been placed on discovering the specific cellular proteins that recognize cisplatin-DNA complexes and then examining how the interaction of these proteins with the complexes might lead to programmed cell death of cancer cells.1
Cellular Uptake of Cisplatin
Before we describe the interactions of cisplatin in the cell, we need to understand how it gets there. Cisplatin is administered to cancer patients intravenously as a sterile saline solution (that is, containing salt—specifically, sodium chloride). Once cisplatin is in the bloodstream, it remains intact due the relatively high concentration of chloride ions (~100 mM). The neutral compound then enters the cell by either passive diffusion or active uptake by the cell. Inside the cell, the neutral cisplatin molecule undergoes hydrolysis, in which a chlorine ligand is replaced by a molecule of water, generating a positively charged species, as shown below and in Figure 1. Hydrolysis occurs inside the cell due to a much lower concentration of chloride ion (~3-20 mM)—and therefore a higher concentration of water.
inside the cell: PtII(NH3)2Cl2 + H2O -> [ PtII(NH3)2Cl(H2O)]+ + Cl-
[PtII(NH3)2Cl(H2O)]+ + H2O -> [PtII(NH3)2(H2O)2]2+
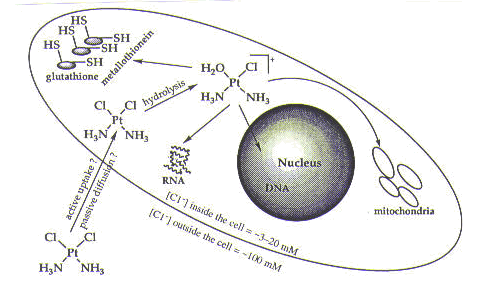
As seen in Figure 1, once inside the cell, cisplatin has a number of possible targets: DNA; RNA; sulfur-containing enzymes such as metallothionein and glutathione; and mitochondria. The effects of DNA on mitochondria are not well understood, but it is possible that damage to mitochondrial DNA resulting from cisplatin treatment contributes to cell death. The interaction of cisplatin with sulfur-containing enzymes is better understood and is believed to be involved in resistance of cells to cisplatin (as discussed in the drug resistance module). The effects of cisplatin on RNA and DNA have been studied extensively. We will briefly discuss the interaction of cisplatin with RNA. We will also describe the interaction of cisplatin with DNA in some detail and discuss why DNA is the target of cisplatin that is believed to be responsible for cell death.1
Interaction of Cisplatin with RNA
Although cisplatin can coordinate to RNA, this interaction is not believed to play an important role in cisplatin’s mechanism of action in the body for two reasons. First, a single damaged RNA molecule can be replaced by newly synthesized material; studies have revealed that cisplatin does not affect RNA synthesis (but that it does affect DNA synthesis). Second, when cisplatin was administered in vitro at its lethal dose to a strain of cancer cells, only a small fraction (1% to 10%) of RNA molecules were damaged.1
Interaction of Cisplatin with DNA
Cisplatin coordinates to DNA mainly through certain nitrogen atoms of the DNA base pairs; these nitrogen atoms (specifically, the N7 atoms of purines) are free to coordinate to cisplatin because they do not form hydrogen bonds with any other DNA bases, as shown in Figure 2. (Also see the module on DNA.)
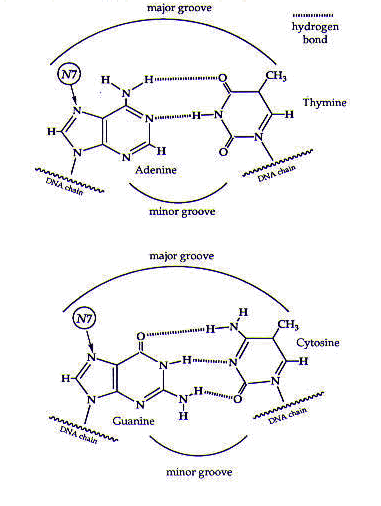
Figure 2. The DNA base pairs. Cisplatin coordinates to the N7 atoms of the purine (guanine and adenine) bases. Reprinted with permission.1
Many types of cisplatin–DNA coordination complexes, or adducts, can be formed. The most important of these appear to be the ones in which the two chlorine ligands of cisplatin are replaced by purine nitrogen atoms on adjacent bases on the same strand of DNA; these complexes are referred to 1,2-intrastrand adducts. The purine bases most commonly involved in these adducts are guanines; however, adducts involving one guanine and one adenine are also found. The formation of these adducts causes the purines to become destacked and the DNA helix to become kinked, as shown in Figure 3.
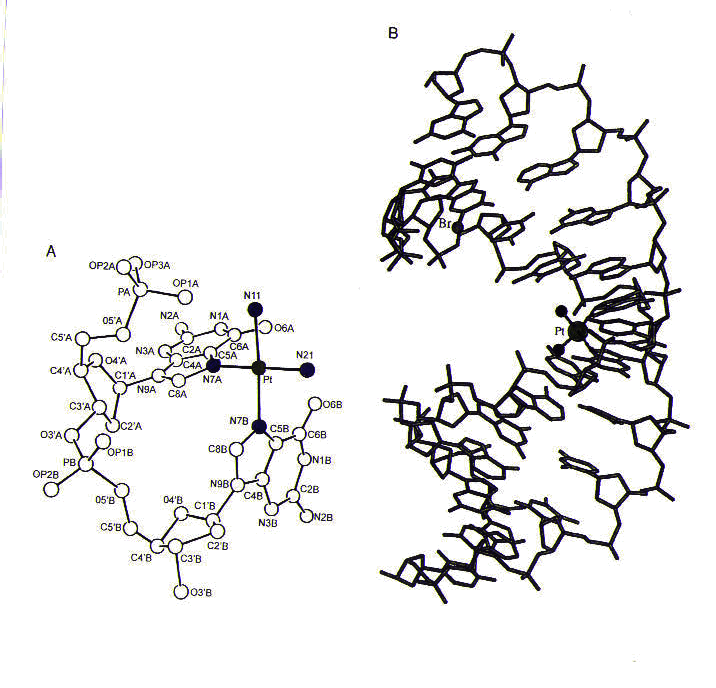
Figure 3. (a) The structure of cisplatin (with platinum shown in green and nitrogen ligands shown in blue) coordinated to a dinucleotide containing two guanines. Notice the destacking of guanine bases, which would normally be parallel to one another. (b) The structure of cisplatin coordinated to two guanines in a DNA duplex. Reprinted with permission.1
Due to its geometry, trans-DDP cannot form 1,2-intrastrand adducts with DNA. Since trans-DDP is inactive in killing cancer cells, it is believed that the 1,2-intrastrand adducts formed between cisplatin and DNA are important for the anticancer activity of cisplatin.1
We have seen how cisplatin binds DNA, and now we want to understand how the binding of cisplatin to DNA leads to programmed cell death. Researchers have found that this binding affects both replication and transcription of DNA, as well as mechanisms of DNA repair. The effects of both cisplatin and trans-DDP on DNA replication were studied both in vitro (using cell extracts outside the host organism) and in vivo (inside the host organism). In vitro studies on both prokaryotic (bacterial) and eukaryotic (mammalian) cells revealed that DNA adducts of both cisplatin and trans-DDP blocked the action of DNA polymerase, an enzyme necessary for replication. In particular, 1,2-intrastrand adducts of cisplatin with DNA all stopped polymerases from doing their job. Likewise, in vivo studies showed that cisplatin and trans-DDP inhibited replication equally well. Since other studies have shown that cisplatin is an effective antitumor agent but trans-DDP is not, these results suggest that DNA replication is not the only factor important for the clinical activity of cisplatin in destroying cancer cells.1 The effects of cisplatin and trans-DDP on DNA transcription are harder to interpret than the effects on replication. However, cisplatin does not appear to inhibit transcription, possibly leading to programmed cell death.1
Cisplatin and DNA Repair
The cytotoxic activity of cisplatin may arise from the cell’s inability to repair DNA damage caused by cisplatin. Indeed, in vitro studies on cell extracts suggest that the most common cisplatin–DNA adducts (that is, 1,2-intrastrand adducts) are not readily repaired by the excision repair system (see DNA module). It is dangerous to draw too many conclusions from these studies, however, because there may be mechanisms of repair present in the organism that are not apparent from studies on cell extracts alone.1
Interactions of Cellular Proteins with Cisplatin-Damaged
DNA Researchers have conducted further studies to address the possibility that cisplatin’s cytotoxic activity may result from a failure of the excision repair system. In this repair system before the damaged portion of DNA is even excised from the rest of the strand, it must be recognized by the cell. The cell detects DNA damage by the action of damage recognition proteins. Therefore, as a first step in studying the excision repair system, researchers looked for evidence of proteins attached to cisplatin–DNA adducts. Several types of assays can differentiate between DNA that is bound to a protein and free DNA; researchers have been able to use these assays to isolate several proteins that bind to cisplatin–DNA adducts. These proteins all contain a common portion (that is, similar or even identical sequences of amino acids, which are the building blocks of proteins) called a high mobility group (HMG); proteins in this class are called HMG-domain proteins. The tests described above have shown that HMG-domain proteins bind cisplatin–DNA adducts in vitro . In vivo assays on yeast have also provided evidence that HMG-domain proteins are important for the activity of cisplatin: cells lacking the gene that codes for HMG-domain proteins are less sensitive to cisplatin than cells containing the gene, meaning that cisplatin is less effective in killing these cells. This result suggests that HMG-domain proteins play an important role in cisplatin’s activity in killing cells; these effects may also be in operation in mammalian cells.1 Two theories explain the possible role of HMG-domain proteins in cisplatin’s cytotoxic activity. Many HMG-domain proteins are transcription factors, meaning that they are required for the synthesis of RNA from a DNA template. One theory asserts that if HMG-domain-containing transcription factors bind preferentially to the cisplatin–DNA adducts, they could wreak havoc with the transcriptional machinery, possibly leading to cell death. A second theory suggests that when HMG-domain proteins bind to the cisplatin–DNA adducts, the adducts would not be recognized by the repair machinery.2 DNA repair would then be slower than normal, as shown in Figure 4.
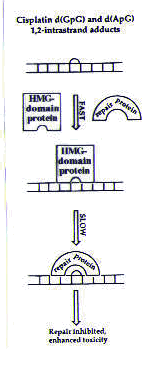 |
Figure 4. Model for the inhibition of cisplatin adduct repair in the presence of HMG-domain proteins. Reprinted with permission.1 |
The cisplatin–DNA adducts would then be more persistent than they would in the absence of HMG-domain proteins, and DNA repair would be slower. This could interfere with the normal functions of the cell (among them, replication and transcription) and possibly trigger cell death.2
For some recent developments in this area, go to the module on further studies.
References
- Pil, P., Lippard, S. J. In Encyclopedia of Cancer, J. R. Bertino, Ed. Academic Press: San Diego, CA, 1997, Vol. 1, pp. 392-410.
- Marla, S., Personal communication, 1999.