
17.6: Catalysts and Catalysis
- Page ID
- 46073

Make sure you thoroughly understand the following essential ideas which have been presented above. It is especially imortant that you know the precise meanings of all the green-highlighted terms in the context of this topic.
- What are catalysts, and how do they work in terms altering the parameters of a reaction?
- Describe the similarities and differences between the three principal classes of catalysts.
- Define physisorption and chemisorption, and explain the role of the latter in initiating a catalytic event.
- What is the meaning and significance of the Sabatier Principle?
- What are the principal differences between the Langmuir-Hinshelwood and Eley-Rideal mechanisms of heterogeneous catalysis?
- Describe the role of the enzyme-substrate complex.
- How might the particular properties of the amino-acids surrounding the active site of an enzyme contribute to its catalytic effect?
- Describe the lock-and-key and induced-fit models of enzyme action.
- Explain the function and significance of allosteric sites on an enzyme.
It almost seems like magic! A mixture of gaseous H2 and O2 can coexist indefinitely without any detectable reaction, but if a platinum wire is heated and then immersed in the gaseous mixture, the reaction proceeds gently as the heat liberated by the reaction makes the wire glow red-hot. Catalysts play an essential role in our modern industrial economy, in our stewardship of the environment, and in all biological processes. This lesson will give you a glimpse into the wonderful world of catalysts, helping you to understand what they are and how they work.
What are Catalysts?
Catalysts have no effect on the equilibrium constant and thus on the equilibrium composition. Catalysts are substances that speed up a reaction but which are not consumed by it and do not appear in the net reaction equation. Also — and this is very important — catalysts affect the forward and reverse rates equally; this means that catalysts have no effect on the equilibrium constant and thus on the composition of the equilibrium state. Thus a catalyst (in this case, sulfuric acid) can be used to speed up a reversible reaction such as ester formation or its reverse, ester hydrolysis:
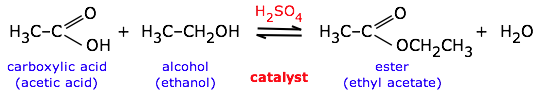
The catalyst has no effect on the equilibrium constant or the direction of the reaction. The direction can be controlled by adding or removing water (Le Chatelier principle).
Catalysts function by allowing the reaction to take place through an alternative mechanism that requires a smaller activation energy. This change is brought about by a specific interaction between the catalyst and the reaction components. You will recall that the rate constant of a reaction is an exponential function of the activation energy, so even a modest reduction of \(E_a\) can yield an impressive increase in the rate.
Catalysts provide alternative reaction pathways
Catalysts are conventionally divided into two categories: homogeneous and heterogeneous. Enzymes, natural biological catalysts, are often included in the former group, but because they share some properties of both but exhibit some very special properties of their own, we will treat them here as a third category.
Some common examples of catalysis
How to burn a Sugar Cube
When heated by itself, a sugar cube (sucrose) melts at 185°C but does not burn. But if the cube is rubbed in cigarette ashes, it burns before melting owing to the catalytic action of trace metal compounds in the ashes.
Platinum as an Oxidation Catalyst
The surface of metallic platinum is an efficient catalyst for the oxidation of many fuel vapors. This property is exploited in flameless camping stoves (left). The image at the right shows a glowing platinum wire heated by the slow combustion of ammonia on its surface. However, if you dip a heated Pt wire into liquid ammonia, you get a miniature explosion: see video below.
This is oxidation of ammonia by oxygen catalysed by warmed platinum wire. Oxygen is bubbled through ammonia solution, in which it mixes with ammonia gas present. The reaction causes the platinum wire to glow, and the hot wire ignites a mixture of ammonia and oxygen.
Decomposition of Hydrogen Peroxide
Hydrogen peroxide is thermodynamically unstable according to the reaction
\[\ce{2 H2O2 → 2 H2O + O2 } \quad \quad ΔG^o = –210\, kJ\, mol^{–1}\]
In the absence of contaminants this reaction is very slow, but a variety of substances, ranging from iodine, metal oxides, trace amount of metals, greatly accelerate the reaction, in some cases almost explosively owing to the rapid release of heat. The most effective catalyst of all is the enzyme catalase, present in blood and intracellular fluids; adding a drop of blood to a solution of 30% hydrogen peroxide induces a vigorous reaction.
Rapid liberation of O2 can result in a spectacular bubble bath if some soap is added. [This same reaction has been used to power a racing car!
Potassium iodide efficiently catalyzes H2O2 deposition. This short video shows what happens when some colored soap, H2O2, and KI are combined. But "don't try this at [your] home"!
Each kind of catalyst facilitates a different pathway with its own activation energy. Because the rate is an exponential function of Ea (Arrhenius equation), even relatively small differences in Ea's can have dramatic effects on reaction rates. Note especially the values for catalase; the chemist is still a rank amateur compared to what Nature can accomplish through natural selection!
| catalyst |
Ea |
relative rate |
|---|---|---|
| no catalyst | 75 | 1 |
| iodide ion | 56 | 2140 |
| colloidal platinum | 50 | 24,000 |
| catalase (enzyme) | 21 | 2,900,000,000 |
How catalytic activity is expressed
Changes in the rate constant or of the activation energy are obvious ways of measuring the efficacy of a catalyst. But two other terms have come into use that have special relevance in industrial applications.
Turnover number
The turnover number (TON) is an average number of cycles a catalyst can undergo before its performance deteriorates (see below). Reported TONs for common industrial catalysts span a very wide range from perhaps 10 to well over 105, which approaches the limits of diffusion transport.
Turnover Frequency
This term, which was originally applied to enzyme-catalyzed reactions, has come into more general use. It is simply the number of times the overall catalyzed reaction takes place per catalyst (or per active site on an enzyme or heterogeneous catalyst) per unit time:is defined as
The number of active sites S on a heterogeneous catalyst is often difficult to estimate, so it is often replaced by the total area of the exposed catalyst, which is usually experimentally measurable. TOFs for heterogeneous reactions generally fall between 10–2 to 102 s–1.
Homogeneous Catalysis
As the name implies, homogeneous catalysts are present in the same phase (gas or liquid solution) as the reactants. Homogeneous catalysts generally enter directly into the chemical reaction (by forming a new compound or complex with a reactant), but are released in their initial form after the reaction is complete, so that they do not appear in the net reaction equation.
Iodine-catalyzed cis-trans isomerization
Unless you are taking an organic chemistry course in which your instructor indicates otherwise, don't try to memorize these mechanisms. They are presented here for the purpose of convincing you that catalysis is not black magic, and to familiarize you with some of the features of catalyzed mechanisms. It should be sufficient for you to merely convince yourself that the individual steps make chemical sense.
You will recall that cis-trans isomerism is possible when atoms connected to each of two doubly-bonded carbons can be on the same (cis) or opposite (trans) sides of the bond. This reflects the fact that rotation about a double bond is not possible.
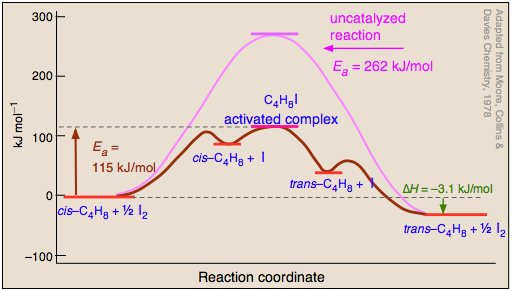
Conversion of an alkene between its cis- and trans forms can only occur if the double bond is temporarily broken, thus freeing the two ends to rotate. Processes that cleave covalent bonds have high activation energies, so cis-trans isomerization reactions tend to be slow even at high temperatures. Iodine is one of several catalysts that greatly accelerate this process, so the isomerization of butene serves as a good introductory example of homogeneous catalysis.
The mechanism of the iodine-catalyzed reaction is believed to involve the attack of iodine atoms (formed by the dissociation equilibrium on one of the carbons in Step
:
During its brief existence, the free-redical activated complex can undergo rotation about the C—C bond, so that when it decomposes by releasing the iodine (), a portion of the reconstituted butene will be in its trans form. Finally, the iodine atom recombine into diiodine. Since processes
and
cancel out, iodine does not appear in the net reaction equation — a requirement for a true catalyst.
Acid-base catalysis
Many reactions are catalyzed by the presence of an acid or a base; in many cases, both acids and bases will catalyze the same reaction. As one might expect, the mechanism involves the addition or removal of a proton, changing the reactant into a more kinetically labile form. A simple example is the addition of iodine to propanone
I2 + (CH3)2–C=O → (CH2I)(CH3)–C=O
The mechanism for the acid-catalyzed process involves several steps. The role of the acid is to provide a proton that attaches to the carbonyl oxygen, forming an unstable oxonium ion . The latter rapidly rearranges into an enol
(i.e., a carbon connected to both a double bond (ene) and a hydroxyl (ol) group.) This completes the catalytic part of the process, which is basically an acid-base (proton-transfer) reaction in which the role of the proton is to withdraw an electron from the ketone oxygen.
In the second stage, the enol reacts with the iodine. The curved arrows indicate shifts in electron locations. In below, an electron is withdrawn from the π orbital of the double bond by one of the atoms of the I2 molecule. This induces a shift of electrons in the latter, causing half of this molecule to be expelled as an iodide ion. The other half of the iodine is now an iodonium ion I+ which displaces a proton from one of the methyl groups. The resultant carbonium ion
then expels the -OH proton to yield the final neutral product.
Perhaps the most well-known acid-catalyzed reaction is the hydrolysis (or formation) of an ester — a reaction that most students encounter in an organic chemistry laboratory course. This is a more complicated process involving five steps; its mechanism is discussed here. See also this U. Calgary site, which describes both the acid- and base-catalyzed reaction.
Oxidation-reduction catalysis
Many oxidation-reduction (electron-transfer) reactions, including direct oxidation by molecular oxygen, are quite slow. Ions of transition metals capable of existing in two oxidation states can often materially increase the rate. An example would be the reduction of Fe3+ by the vanadium ion V3+:
\[\ce{V^{3+} + Fe^{3+} → V^{4+} + Fe^{2+}}\]
This reaction is catalyzed by either Cu+or Cu3+, and the rate is proportional to the concentration of V3+and of the copper ion, but independent of the Fe3+ concentration. The mechanism is believed to involve two steps:
| 1 V3+ + Cu2+ |
(rate-determining) |
| 2 Fe3+ + Cu+ |
(very fast) |
(If Cu+ is used as the catalyst, it is first oxidized to Cu2+ by step 2.)
Hydrogen Peroxide, again
Ions capable of being oxidized by an oxidizing agent such as H2O2 can serve as catalysts for its decomposition. Thus H2O2 oxidizes iodide ion to iodate, when then reduces another H2O2 molecule, returning an I– ion to start the cycle over again:
\[H_2O_2 + I^– → H_2O + IO^–\]
\[H_2O_2 + IO^– → H_2O + O_2 + I^–\]
Iron(II) can do the same thing. Even traces of metallic iron can yield enough Fe2+ to decompose solutions of hydrogen peroxide.
\[H_2O_2 + Fe^{2+} → H_2O + Fe^{3+}\]
\[H_2O_2 + Fe^{3+} + 2H^+ → H_2O + O_2 + Fe^{2+} + 2H^+\]
Heterogeneous catalysts
As its name implies, a heterogeneous catalyst exists as a separate phase (almost always a solid) from the one (most commonly a gas) in which the reaction takes place. The catalytic affect arises from disruption (often leading to dissociation) of the reactant molecules brought about by their interaction with the surface of the catalyst.
← Model of a catalyst consisting of clusters of 8-10 platinum atoms (blue) deposited on an aluminum oxide surface. This catalyst efficiently removes hydrogen atoms from propane, converting it into the industrially-important propylene. [source]
Unbalanced forces at surfaces
You will recall that one universal property of matter is the weak attractive forces that arise when two particles closely approach each other. (See here for a quick review.) When the particles have opposite electric charges or enter into covalent bonding, these far stronger attraction dominate and define the "chemistry" of the interacting species.
The molecular units within the bulk of a solid are bound to their neighbors through these forces which act in opposing directions to keep each under a kind of "tension" that restricts its movement and contributes to the cohesiveness and rigidity of the solid.
At the surface of any kind of condensed matter, things are quite different. The atoms or molecules that reside on the surface experience unbalanced forces which prevents them from assuming the same low potential energies that characterize the interior units. (The same thing happens in liquids, and gives rise to a variety of interfacial effects such as surface tension.)
But in the case of a solid, in which the attractive forces tend to be stronger, something much more significant happens. The molecular units that reside on the surface can be thought of as partially buried in it, with their protruding parts (and the intermolecular attractions that emerge from them) exposed to the outer world. The strength of the attractive force field which emanates from a solid surface varies in strength depending on the nature of the atoms or molecules that make up the solid.
Don't confuse adsorption with absorption; the latter refers to the bulk uptake of a substance into the interior of a porous material. At the microscopic level, of course, absorption also involves adsorption. The process in which molecules in a gas or a liquid come into contact with and attach themselves to a solid surface is known as adsorption. Adsorption is almost always an exothermic process and its strength is conventionally expressed by the enthalpy or "heat" of adsorption ΔHads.
Chemisorption and Physisorption
Two general categories of adsorption are commonly recognized, depending on the extent to which the electronic- or bonding structure of the attached molecule is affected. When the attractive forces arise from relatively weak van der Waals interactions, there is little such effect and ΔHads tends to be small. This condition is described as physical adsorption (physisorption). Physisorption of a gas to a surface is energetically similar to the condensation of the gas to a liquid, it usually builds up multiple layers of adsorbed molecules, and it proceeds with zero activation energy.
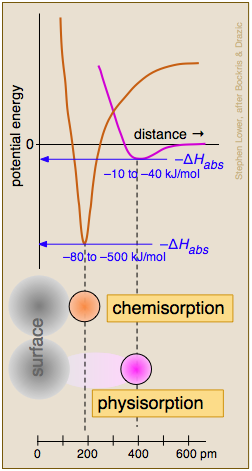
Of more relevance to catalytic phenomena is chemisorption, in which the adsorbate is bound to the surface by what amounts to a chemical bond. The resulting disruption of the electron structure of the adsorbed species "activates" it and makes it amenable to a chemical reaction (often dissociation) that could not be readily achieved through thermal activation in the gas or liquid phase. In contrast to physisorption, chemisorption generally involves an activation energy (supplied by ΔHads) and the adorbed species is always a monolayer.
Mechanisms of reactions on surfaces
Dissociative adsorption
The simplest heterogeneous process is chemisorption followed by bond-breaking as described above. The most common and thoroughly-studied of these is the dissociation of hydrogen which takes place on the surface of most transition metals. The single 1s electron of each hydrogen atom coordinates with the d orbitals of the metal, forming a pair of chemisorption bonds (indicated by the red dashed lines). Although these new bonds are more stable than the single covalent bond they replace, the resulting hydrogen atoms are able to migrate along the surface owing to the continuous extent of the d-orbital conduction band.
The Langmuir-Hinshelwood mechanism
Although the adsorbed atoms ("adatoms") are not free radicals, they are nevertheless highly reactive, so if a second, different molecular species adsorbs onto the same surface, an interchange of atoms may be possible. Thus carbon monoxide can be oxidized to CO2 by the process illustrated below:
In this example, only the O2 molecule undergoes dissociation . The CO molecule adsorbs without dissociation
![]() , configured perpendicular to the surface with the chemisorption bond centered over a hollow space between the metal atoms. After the two adsorbed species have migrated near each other
, configured perpendicular to the surface with the chemisorption bond centered over a hollow space between the metal atoms. After the two adsorbed species have migrated near each other , the oxygen atom switches its attachment from the metal surface to form a more stable C=O bond with the carbon
, followed by release of the product molecule.
The Eley-Rideal mechanism
An alternative mechanism eliminates the second chemisorption step; the oxygen adatoms react directly with the gaseous CO molecules by replacing the chemisorption bond with a new C–O bond as they swoop over the surface:
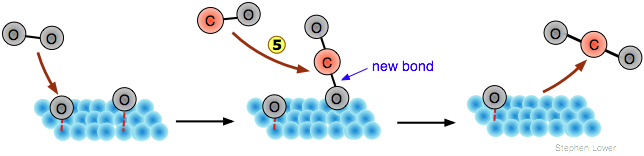
Examples of both mechanisms are known, but the Langmuir-Hinshelwood mechanism is more importantin that it exploits the activation of the adsorbed reactant. In the case of carbon monoxide oxdation, studies involving molecular beam experiments support this scheme. A key piece of evidence is the observation of a short time lag between contact of a CO molecule with the surface and release of the CO2, suggesting that CO remains chemisorbed during the interval.
The Sabatier Principle
To be effective, these processes of adsorption, reaction, and desorption must be orchestrated in a way that depends critically on the properties of the catalyst in relation to the chemisorption properties (ΔHads) of the reactants and products.
- Adsorption of the reactant onto the catalytic surface (2) must be strong enough to perturb the bonding within the species to dissociate or activate it;
- If the resulting fragments must migrate to other locations on the surface (3-4), their chemisorption must be weak enough to allow this movement but not so small that they escape before they have a chance to react;
- The product species must have sufficiently small ΔHads values to ensure their rapid desorption from the catalyst (5) so that surface is freed up to repeat the cycle
The importance of choosing a catalyst that achieves the proper balance of the heats of adsorption of the various reaction components is known as the Sabatier Principle, but is sometimes referred to as the "just-right" or "Goldilocks principle". Remember the story of Goldilocks and the Three Bears? ... or see this UTube video.
In its application to catalysis, this principle is frequently illustrated by a "volcano diagram" in which the rate of the catalyzed reaction is plotted as a function of ΔHads of a substrate such as H2 on a transition metal surface.
The plot at the left shows the relative effectiveness of various metals in catalyzing the decomposition of formic acid HCOOH. The vertical axis is plotted as temperature, the idea being that the better the catalyst, the lower the temperature required to maintain a given rate.
The catalytic cycle
This term refers to the idealized sequence of steps between the adsorption of a reactant onto the catalyst and the desorption of the product, culminating in restoration of the catalyst to its original condition. A typical catalytic cycle for the hydrogenation of propene is illustrated below.
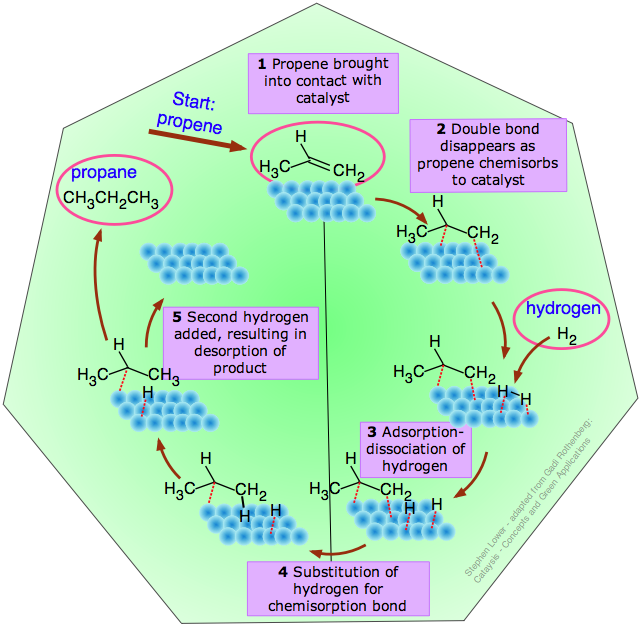
This particular reaction
H3C–CH=CH2 + H2 → H3C–CH2–CH3
takes place spontaneously only in the reverse direction, but it is representative of the process used to hydrogenate the carbon-carbon double bonds in vegetable oils to produce solid saturated fats such as margarine.
Catalyst poisoning and breakdown
Catalyst poisoning, brought about by irreversible binding of a substance to its surface, can be permanent or temporary. In the latter case the catalyst can be regenerated, usually by heating to a high temperature. In organisms, many of the substances we know as "poisons" act as catalytic poisons on enzymes. If catalysts truly remain unchanged, they should last forever, but in actual practice, various events can occur that limit the useful lifetime of many catalysts.
- Impurities in the feedstock or the products of side reactions can bind permanently to a sufficient number of active sites to reduce catalytic efficiency over time, "poisoning" the catalyst.
- Physical deterioration of the catalyst or of its support, often brought about by the high temperatures sometimes used in industrial processes, can reduce the effective surface area or the accessibility of reactants to the active sites.
Catalysts tend to be rather expensive, so it is advantageous if they can be reprocessed or regenerated to restore their activity. It is a common industrial practice to periodically shut down process units to replace spent catalysts.
How heterogeneous catalysts work
The actual mechanisms by which adsorption of a molecule onto a catalytic surface facilitates the cleavage of a bond vary greatly from case to case.
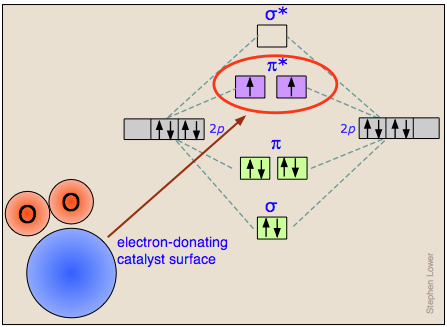
We give here only one example, that of the dissociation of dixoygen O2 on the surface of a catalyst capable of temporarily donating an electron which enters an oxygen antibonding molecular orbital that will clearly destabilize the O–O bond. (Once the bond has been broken, the electron is given back to the catalyst.)
Types of catalytically active surfaces
Heterogeneous catalysts mostly depend on one or more of the followng kinds of surfaces:
| Active surface type | Remarks |
|---|---|
| Atoms at surfaces or crystal edges in macromolecular 2- and 3-D networks such as graphite or quartz may have free valences | |
| Surfaces and edges are sites of intense electric fields able to interact with ions and polar molecules | |
| Oxides can acquire H+ and/or OH– groups able to act as acid- or base catalyts | |
| "Electron gas" at metal surfaces can perturb bonding in substrate molecules | |
 |
Vacant d orbitals can provide a variety of coordination sites for activation. |
| Semiconductors (including many oxides) can supply electrons, thermally excited through reasonably small (<50 kJ) band gaps. |
Some factors affecting catalyst efficacy
Since heterogeneous catalysis requires direct contact between the reactants and the catalytic surface, the area of active surface goes at the top of the list. In the case of a metallic film, this is not the same as the nominal area of the film as measured by a ruler; at the microscopic level, even apparently smooth surfaces are highly irregular, and some cavities may be too small to accommodate reactant molecules.
Consider, for example, that a 1-cm cube of platinum (costing roughly $1000) has a nominal surface area of only 6 cm2. If this is broken up into 1012 smaller cubes whose sides are 10–6 m, the total surface area would be 60,000 cm2, capable in principle of increasing the rate of a Pt-catalyzed reaction by a factor of 104. These very finely-divided (and often very expensive) metals are typically attached to an inert supporting surface to maximize their exposure to the reactants.
Surface topography. At the microscopic level, even an apparently smooth surface is pitted and uneven, and some sites will be more active than others. Penetration of molecules into and out of some of the smaller channels of a porous surface may become rate-limiting.
An otherwise smooth surface will always possess a variety of defects such as steps and corners which offer greater exposure and may be either the only active sites on the surface, or overly active so as to permanently bind to a reactant, reducing the active area of the surface. In one study, it was determined that kink defects constituting just 5 percent of platinum surface were responsible for over 90% of the catalytic activity in a certain reaction.
Steric factors
When chemisorbtion occurs at two or more locations on the reactant, efficient catalysis requires that the spacing of the active centers on the catalytic surface be such that surface bonds can be formed without significant angular distortion.
Thus activation of the ethylene double bond on a nickel surface proceeds efficiently because the angle between the C—Ni bonds and the C—C is close to the tetrahedral value of 109.5° required for carbon sp3 hybrid orbital formation. Similarly, we can expect that the hydrogenation of benzene should proceed efficiently on a surface in which the active sites are spaced in the range of 150 and 250 pm.
This is one reason why many metallic catalysts exhibit different catalytic activity on different crystal faces.
Some special types of heterogeneous catalysts
Metal-cluster catalysts
As the particle size of a catalyst is reduced, the fraction of more highly exposed step, edge, and corner atoms increases. An extreme case occurs with nano-sized (1-2 nm) metal cluster structures composed typically of 10-50 atoms.
[link] →
Metallic gold, well known for its chemical inertness, exhibits very high catalytic activity when it is deposited as metallic clusters on an oxide support. For example, O2 dissociates readily on Au55 clusters which have been found to efficiently catalyze the oxidation of hydrocarbons [article].
Zeolite catalysts
Zeolites are clay-like aluminosilicate solids that form open-framework microporous structures that may contain linked cages, cavities or channels whose dimensions can be tailored to the sizes of the reactants and products. To those molecules able to diffuse through these spaces, zeolites are in effect "all surface", making them highly efficient. This size-selectivity makes them important for adsorption, separation, ion-exchange, and catalytic applications. Many zeolites occur as minerals, but others are made synthetically in order to optimize their properties.
As catalyts, zeolites offer a number of advantages that has made them especially important in "green chemistry" operations in which the number of processing steps, unwanted byproducts, and waste stream volumes are minimized.
Enzymes and biocatalysis
"A Jug of Wine, a Loaf of Bread, and . . ." Enzymes!
This distortion of Robert FitzGerald's already-distorted translation of the famous quatrain from the wonderful Rubaiyat of Omar Khayyam underlines the central role that enzymes and their technology have played in civilization since ancient times.
← Illustration by Edmund Dulac (1882-1953)
Fermentation and wine-making have been a part of human history and culture for at least 8000 years, but recognition of the role of catalysis in these processes had to wait until the late nineteenth century. By the 1830's, numerous similar agents, such as those that facilitate protein digestion in the stomach, had been discovered. The term "enzyme", meaning "from yeast", was coined by the German physiologist Wilhelm Kühne in 1876. In 1900, Eduard Buchner (1860-1917, 1907 Nobel Prize in Chemistry) showed that fermentation, previously believed to depend on a mysterious "life force" contained in living organisms such as yeast, could be achieved by a cell-free "press juice" that he squeezed out of yeast.
By this time it was recognized that enzymes are a form of catalyst (a term introduced by Berzelius in 1835), but their exact chemical nature remained in question. They appeared to be associated with proteins, but the general realization that enzymes are proteins began only in the 1930s when the first pure enzyme was crystallized, and did not become generally accepted until the 1950s. It is now clear that nearly all enzymes are proteins, the major exception being a small but important class of RNA-based enzymes known as ribozymes.
Proteins are composed of long sequences of amino acids strung together by amide bonds; this sequence defines the primary structure of the protein.. Their huge size (typically 200-2000 amino acid units, with total molecular weights 20,000 - 200,000) allows them to fold in complicated ways (known as secondary and tertiary structures) whose configurations are essential to their catalytic function.
Because enzymes are generally very much larger than the reactant molecules they act upon (known in biochemistry as substrates), enzymatic catalysis is in some ways similar to heterogeneous catalysis. The main difference is that the binding of a subtrate to the enzyme is much more selective.
Precursors and cofactors
Most enzymes come into being as inactive precursors (zymogens) which are converted to their active forms at the time and place they are needed.
- For example, the enzymes that lead to the clotting of blood are supposed to remain inactive until bleeding actually begins; a major activating factor is exposure of the blood to proteins in the damaged vessel wall.
- The enzyme that catalyzes the formation of lactose (milk sugar) in the mammary gland is formed during pregnancy, but it remains inactive until the time of birth, when hormonal changes cause a modifier unit to bind to and activate the enzyme.
Conversion to the active form may involve a simple breaking up of the protein by hydrolysis of an appropriate peptide bond or the addition of a phosphate or similar group to one of the amino acid residues.
Many enzyme proteins also require "helper" molecules, known as cofactors, to make them catalytically active. These may be simple metal ions (many of the trace nutrient ions of Cu, Mn, Mo, V, etc.) or they may be more complex organic molecules which are called coenzymes. Many of the latter are what we commonly refer to as vitamins. Other molecules, known as inhibitors, decrease enzyme activity; many drugs and poisons act in this way.
The enzyme-substrate complex
The standard model of enzyme kinetics consists of a two-step process in which an enzyme binds reversibly to its substrate S (the reactant) to form an enzyme-substrate complex ES:
The enzyme-substrate complex plays a role similar to that of the activated complex in conventional kinetics, but the main function of the enzyme is to stabilize the transition state.
In the second, essentially irreversible step, the product and the enzyme are released:
The basic kinetic treatment of this process involves the assumption that the concentrations [E] and [ES] reach steady-state values which do not change with time. (The detailed treatment, which is beyond the scope of this course, can be found here.)
The overall process is described by the Michaelis-Menten equation which is plotted here. The Michaelis constant kM is defined as shown, but can be simplified to the ES dissociation constant k–1/k1 in cases when dissociation of the complex is the rate-limiting step. The quantity vmax is not observed directly, but can be determined from kM as shown here.
Enzymes are proteins
In order to understand enzymes and how they catalyze reactions, it is first necessary to review a few basic concepts relating to proteins and the amino acids of which they are composed.
Amino acids: polar, nonpolar, positive, negative.
The 21 amino acids that make up proteins all possess the basic structure shown here, where R represents either hydrogen or a side chain which may itself contain additional –NH2 or –COOH groups. Both kinds of groups can hydrogen-bond with water and with the polar parts of substrates, and therefore contribute to the amino acid's polarity and hydophilic nature. Side chains that contain longer carbon chains and especially benzene rings have the opposite effect, and tend to render the amino acid non-polar and hydrophobic.
Both the –NH2 and –COOH groups are ionizable (i.e., they can act as proton donors or acceptors) and when they are in their ionic forms, they will have an electric charge. The –COOH groups have pKa's in the range 1.8-2.8, and will therefore be in their ionized forms –COO– at ordinary cellular pH values of around 7.4. The amino group pKa's are around 8.8-10.6, so these will also normally be in their ionized forms NH3+.
This means that at ordinary cellular pH, both the carboxyl and amino groups will be ionized. But because the charges have opposite signs, an amino acid that has no extra ionizable groups in its side chain will have a net charge of zero. But if the side chain contains an extra amino or carboxyl group, the amino acid can carry a net electric charge. The following diagram illustrates typical amino acids that fall into each of the groups described above.
Proteins
Proteins are made up of one or more chains of amino acids linked to each other through peptide bonds by elimination of a water molecule.
The product shown above is called a peptide, specifically it is a dipeptide because it contains two amino acid residues (what's left after the water has been removed.) Proteins are simply very long polypeptide chains, or combinations of them. (The distinction between a long polypeptide and a small protein is rather fuzzy!)
Globular proteins
Most enzymes fall into the category of globular proteins. In contrast to the fibrous proteins that form the structural components of tissues, globular proteins are soluble in water and rarely have any systematic tertiary structures. They are made up of one or more amino-acid ("peptide") chains which fold into various shapes that can roughly be described as spherical — hence the term "globular", and the suffix "globin" that is frequently appended to their names, as in "hemoglobin".
Protein folding is a spontaneous process that is influenced by a number of factors. One of these is obviously their primary amino-acid sequence that facilitates formation of intramolecular bonds between amino acids in different parts of the chain. These consist mostly of hydrogen bonds, although disulfide bonds S—S between sulfur-containing amino acids are not uncommon.
In addition to these intramolecular forces, interactions with the surroundings play an important role. The most import of these is the hydrophobic effect, which favors folding conformations in which polar amino acids (which form hydrogen bonds with water) are on the outside, while the so-called hydrophobic amino acids remain in protected locations within the folds.
How enzymes work: the active site
The catalytic process mediated by an enzyme takes place in a depression or cleft that exposes the substrate to only a few of the hundreds-to-thousands of amino acid residues in the protein chain. The high specificity and activity of enzyme catalysis is sensitively dependent on the shape of this cavity and on the properties of the surrounding amino acids
In 1894, long before it was clear that enzymes are proteins, the German chemist Emil Fischer suggested the so-called lock-and-key model as a way of understanding how a given enzyme can act specificilly on only one kind of substrate molecule. This model is essentially an elaboration of the one we still use for explaining heterogeneous catalysis.
Although the basic lock-and-key model continues to be useful, it has been modified into what is now called the induced-fit model. This assumes that when the substrate enters the active site and interacts with the surrounding parts of the amino acid chain, it reshapes the active site (and perhaps other parts of the enzyme) so that it can engage more fully with the substrate.
One important step in this process is to squeeze out any water molecules that are bound to the substrate and which would interfere with its optimal positioning.
Within the active site, specific interactions between the substrate and appropriately charged, hydrophlic and hydrophobic amino acids of the active site then stabilize the transition state by distorting the substrate molecule in such a way as to lead to a transition state having a substantially lower activation energy than can be achieved by ordinay non-enzymatic catalysis. Beyond this point, the basic catalytic steps are fairly conventional, with acid/base and nucleophilic catalysis being the most common.
For a very clear and instructive illustration of a typical multi-step sequence of a typical enzyme-catalyzed reaction, see this page from Mark Bishop's online textbook, from which this illustration is taken.
Enzyme regulation and inhibition
If all the enzymes in an organism were active all the time, the result would be runaway chaos. Most cellular processes such as the production and utilization of energy, cell division, and the breakdown of metabolic products must operate in an exquisitely choreographed, finely-tuned manner, much like a large symphony orchestra; no place for jazz-improv here!
Nature has devised various ways of achieving this; we described the action of precursors and coenzymes above. Here we focus on one of the most important (and chemically-interesting) regulatory mechanisms.
Allosteric regulation: tweaking the active site
There is an important class of enzymes that possess special sites (distinct from the catalytically active sites) to which certain external molecules can reversibly bind. Although these allosteric sites, as they are called, may be quite far removed from the catalytic sites, the effect of binding or release of these molecules is to trigger a rapid change in the folding pattern of the enzyme that alters the shape of the active site. The effect is to enable a signalling or regulatory molecule (often a very small one such as NO) to modulate the catalytic activity of the active site, effectively turning the enzyme on or off.
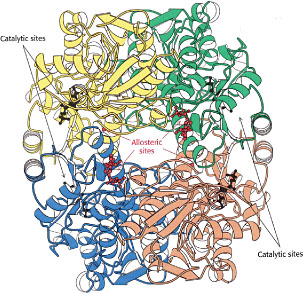
In some instances, the product of an enzyme-catalyzed reaction can itself bind to an allosteric site, decreasing the activity of the enzyme and thus providing negative feedback that helps keep the product at the desired concentration. It is believed that concentrations of plasma ions such as calcium, and of energy-supplying ATP are, are regulated in this way.
Allosteric enzymes are more than catalysts: they act as control points in metabolic and cellular signalling networks.Allosteric enzymes frequently stand at the beginning of a sequence of enzymes in a metabolic chain, or at branch points where two such chains diverge, acting very much like traffic signals at congested intersections.
Enzyme inhibition
As is the case with heterogeneous catalysts, certain molecules other than the normal substrate may be able to enter and be retained in the active site so as to competitively inhibit an enzyme's activity. This is how penicillin and related antibiotics work; these molecules covalently bond to amino acid residues in the active site of an enzyme that catalyzes the formation of an essential component of bacterial cell walls. When the cell divides, the newly-formed progeny essentially fall apart.
Enzymes outside the cell
Enzymes have been widely employed in the food, pulp-and-paper, and detergent industries for a very long time, but mostly as impure whole-cell extracts.
In recent years, developments in biotechnology and the gradual move of industry from reliance on petroleum-based feedstocks and solvents to so-called "green" chemistry have made enzymes more attractive as industrial catalysts. Compared to the latter, purified enzymes tend to be expensive, difficult to recycle, and unstable outside of rather narrow ranges of temperature, pH, and solvent composition.
Immobilized enzymes
Many of the problems connected with the use of free enzymes can be overcome by immobilizing the enzyme. This can be accomplished in several ways:
- Binding the enzyme to an inert solid supporting material
This was first done in 1916, using charcoal or aluminum hydroxide. since the 1960's when this method became more popular, synthetic polymers, often engineered for a specific application, have come into wide use. Some natural biopolymers such as cellulose or starch, and inorganic solids such as silica and alumina have also been employed.
- Trapping the enzyme in a porous material
- Cross-linked enzymes
Catalysis
- Gadi Rothenberg- Catalysis: Concepts and Green Applications (Wiley-VCH, 2008)
- R.A. van Santan, M. Neurock - Molecular Heterogeneous Catalysis: A conceptual and computational approach (Wiley-VCH, 2006)
- History of Catalysis: List of Web pages, including Fifty Years of Catalysis - A list of major advances in the field 1949-1999. More detailed History pages from UK
- Catalysis and its applications - a very readable and interesting account of the applications of catalysis to various industrially-important fields.