
Alkenes from Aldehydes and Ketones - Wittig Reaction
- Page ID
- 66756
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide (often called a Wittig reagent) to give an alkene and triphenylphosphine oxide.

The Wittig reaction was discovered in 1954 by Georg Wittig, for which he was awarded the Nobel Prize in Chemistry in 1979. It is widely used in organic synthesis for the preparation of alkenes. It should not be confused with the Wittig rearrangement.
Wittig reactions are most commonly used to couple aldehydes and ketones to singly substituted phosphine ylides. With unstabilised ylides this results in almost exclusively the Z-alkene product. In order to obtain the E-alkene, stabilised ylides are used or unstabilised ylides using the Schlosser modification of the Wittig reaction can be performed.
Reaction mechanism
Classical mechanism
The steric bulk of the ylide 1 influences the stereochemical outcome of nucleophilic addition to give a predominance of the betaine 3 (cf. Bürgi–Dunitz angle). Note that for betaine 3 both R1 and R2 as well as PPh3+ and O− are positioned anti to one another.
Carbon-carbon bond rotation gives the betaine 4, which then forms the oxaphosphetane 5. Elimination gives the desired Z-alkene 7 and triphenylphosphine oxide 6. With simple Wittig reagents, the first step occurs easily with both aldehydes and ketones, and the decomposition of the betaine (to form 5) is the rate-determining step. However, with stabilised ylides (where R1 stabilises the negative charge) the first step is the slowest step, so the overall rate of alkene formation decreases and a bigger proportion of the alkene product is the E-isomer. This also explains why stabilised reagents fail to react well with sterically hindered ketones.

Mechanism
Mechanistic studies have focused on unstabilized ylides, because the intermediates can be followed by NMR spectroscopy. The existence and interconversion of the betaine (3a and 3b) is subject of ongoing research. Phosphonium ylides 1 react with carbonyl compounds 2 via a π²s/π²a [2+2] cycloaddition to directly form the oxaphosphetanes 4a and 4b. The stereochemistry of the product 5 is due to the addition of the ylide 1 to the carbonyl 2 and to the equilibration of the intermediates. Maryanoff and Reitz identified the issue about equilibration of Wittig intermediates and termed the process "stereochemical drift". For many years, the stereochemistry of the Wittig reaction, in terms of carbon-carbon bond formation, had been assumed to correspond directly with the Z/E stereochemistry of the alkene products. However, certain reactants do not follow this simple pattern. Lithium salts can also exert a profound effect on the stereochemical outcome.

Mechanisms differ for aliphatic and aromatic aldehydes and for aromatic and aliphatic phosphonium ylides. Evidence suggests that the Wittig reaction of unbranched aldehydes under lithium-salt-free conditions do not equilibrate and are therefore under kinetic reaction control. Vedejs has put forth a theory to explain the stereoselectivity of stabilized and unstabilized Wittig reactions.
Wittig reagents
Preparation of phosphorus ylides
Wittig reagents are usually prepared from a phosphonium salt, which is in turn prepared by the quaternization of triphenylphosphine with an alkyl halide. The alkylphosphonium salt is deprotonated with a strong base such as n-butyllithium:
- [Ph3P+CH2R]X− + C4H9Li → Ph3P=CHR + LiX + C4H10
One of the simplest ylides is methylenetriphenylphosphorane (Ph3P=CH2). It is also a precursor to more elaborate Wittig reagents. Alkylation of Ph3P=CH2 with a primary alkyl halide R−CH2−X, produces substituted phosphonium salts:
- Ph3P=CH2 + RCH2X → Ph3P+CH2CH2R X−
These salts can be deprotonated in the usual way to give Ph3P=CH−CH2R.
Structure of the ylide
 Ball-and-stick model of Ph3P=CH2, as found in the crystal structure
Ball-and-stick model of Ph3P=CH2, as found in the crystal structureThe Wittig reagent may be described in the phosphorane form (the more familiar representation) or the ylide form:

The ylide form is a significant contributor, and the carbon is nucleophilic.
Reactivity
Simple phosphoranes are reactive. Most hydrolyze and oxidize readily. They are therefore prepared using air-free techniques. Phosphoranes are more air-stable when they contain an electron withdrawing group. Some examples are Ph3P=CHCO2R and Ph3P=CHPh. These ylides are sufficiently stable to be sold commercially 
Scope and limitations
The Wittig reaction is a popular method for the synthesis of alkene from ketones and aldehydes. The Wittig reagent can generally tolerate carbonyl compounds containing several kinds of functional groups such as OH, OR, aromatic nitro and even ester groups. There can be a problem with sterically hindered ketones, where the reaction may be slow and give poor yields, particularly with stabilized ylides, and in such cases the Horner–Wadsworth–Emmons (HWE) reaction (using phosphonate esters) is preferred. Another reported limitation is the often labile nature of aldehydes which can oxidize, polymerize or decompose. In a so-called Tandem Oxidation-Wittig Process the aldehyde is formed in situ by oxidation of the corresponding alcohol.
As mentioned above, the Wittig reagent itself is usually derived from a primary alkyl halide. Quaternization of triphenylphosphine with most secondary halides is inefficient. For this reason, Wittig reagents are rarely used to prepare tetrasubstituted alkenes. However the Wittig reagent can tolerate many other variants. It may contain alkenes and aromatic rings, and it is compatible with ethers and even ester groups. Even C=O and nitrile groups can be present if conjugated with the ylide- these are the stabilised ylides mentioned above. Bis-ylides (containing two P=C bonds) have also been made and used successfully.
One limitation relates to the stereochemistry of the product. With simple ylides, the product is usually mainly the Z-isomer, although a lesser amount of the E-isomer is often formed also – this is particularly true when ketones are used. If the reaction is performed in DMF in the presence of LiI or NaI, the product is almost exclusively the Z-isomer. If the E-isomer is the desired product, the Schlosser modification may be used. With stabilised ylides the product is mainly the E-isomer, and this same isomer is also usual with the HWE reaction.
Schlosser modification

The major limitation of the traditional Wittig reaction is that the reaction proceeds mainly via the erythro betaine intermediate, which leads to the Z-alkene. The erythro betaine can be converted to the threo betaine using phenyllithium at low temperature. This modification affords the E-alkene.
Allylic alcohols can be prepared by reaction of the betaine ylid with a second aldehyde. For example:
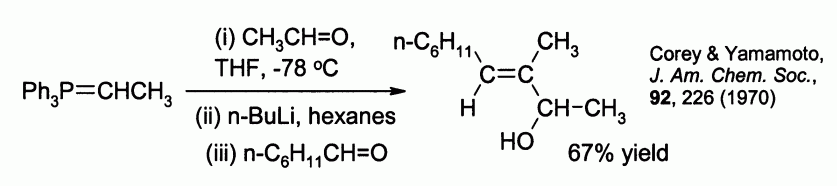
Examples
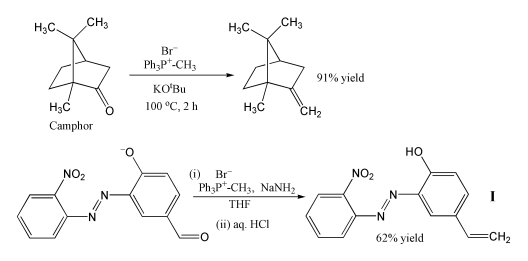
Because of its reliability and wide applicability, the Wittig reaction has become a standard tool for synthetic organic chemists.
The most popular use of the Wittig reaction is for the introduction of a methylene group using methylenetriphenylphosphorane (Ph3P=CH2). Using this reagent even a sterically hindered ketone such as camphor can be converted to its methylene derivative. In this case, the Wittig reagent is prepared in situ by deprotonation of methyltriphenylphosphonium bromide with potassium tert-butoxide. In another example, the phosphorane is produced using sodium amide as a base, and this reagent converts the aldehyde shown into alkene I in 62% yield. The reaction is performed in cold THF, and the sensitive nitro, azo and phenoxide groups are tolerated. The product can be used to incorporate a photostabiliser into a polymer, to protect the polymer from damage by UV radiation.
Another example of its use is in the synthesis of leukotriene A methyl ester. The first step uses a stabilised ylide, where the carbonyl group is conjugated with the ylide preventing self condensation, although unexpectedly this gives mainly the cis product. The second Wittig reaction uses a non-stabilised Wittig reagent, and as expected this gives mainly the cis product. Note that the epoxide and ester functional groups survive intact.
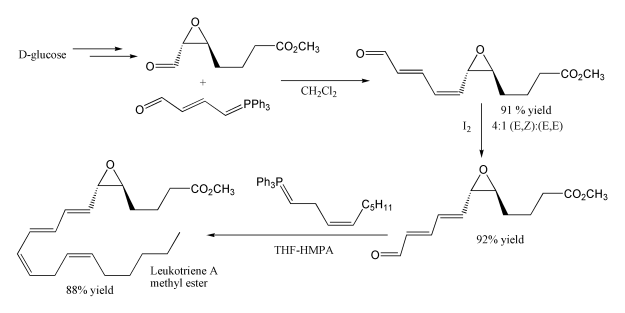
Methoxymethylenetriphenylphosphine is a Wittig reagent for the homologation of aldehydes.