
Additional Methods of Hydroxyl Substitution
- Page ID
- 1168

The most common methods for converting 1º- and 2º-alcohols to the corresponding chloro and bromo alkanes (i.e. replacement of the hydroxyl group) are treatments with thionyl chloride and phosphorus tribromide, respectively. These reagents are generally preferred over the use of concentrated HX due to the harsh acidity of these hydrohalic acids and the carbocation rearrangements associated with their use.
Of course, it is possible to avoid such problems by first preparing a mesylate or tosylate derivative, followed by nucleophilic substitution of the sulfonate ester by the appropriate halide anion. In this two-step approach, a clean configurational inversion occurs in the first SN2 reaction; however, the resulting alkyl halide may then undergo repeated SN2 halogen exchange reactions, thus destroying any stereoisomeric identity held by the initial carbinol carbon. For these and other reasons, alternative mild and selective methods for transforming such alcohols by nucleophilic substitution of the hydroxyl group have been devised. It should be noted that 3º-alcohols are not good substrates for the new procedures.
Disadvantages to using \(PBr_3\) and \(SOCl_2\)
Despite their general usefulness, phosphorous tribromide and thionyl chloride have shortcomings. Hindered 1º- and 2º-alcohols react sluggishly with the former and may form rearrangement products, as noted in the following equation.
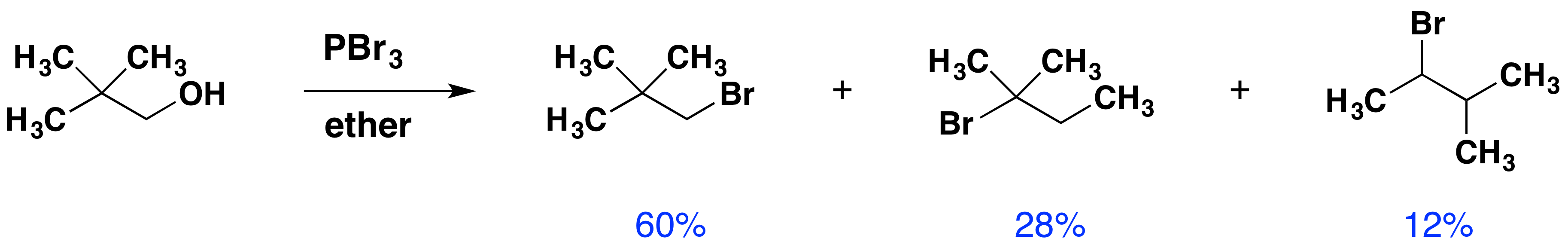
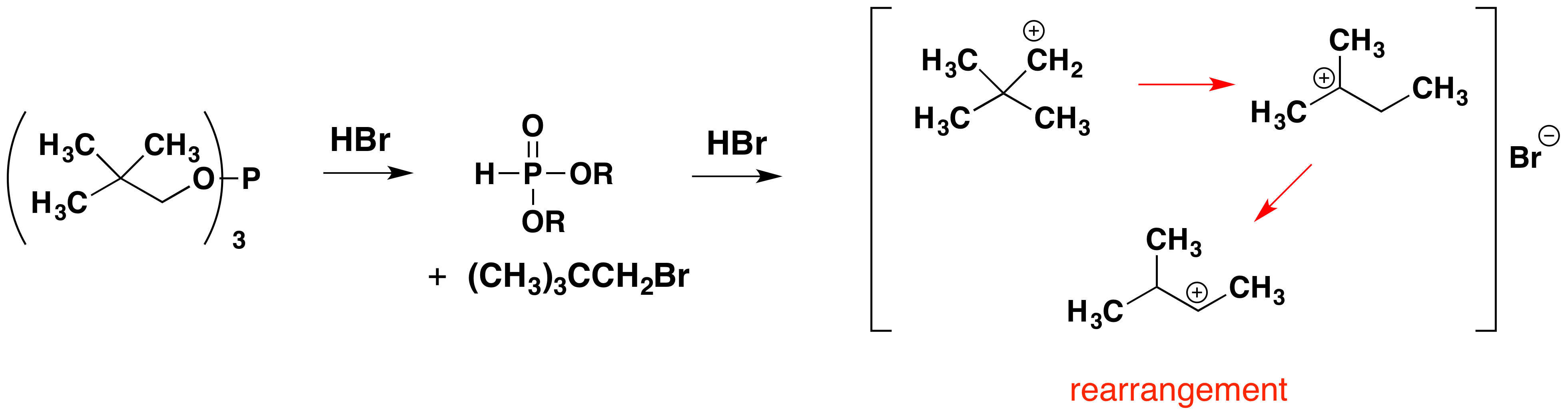
Below, an abbreviated mechanism for the reaction is displayed. The initially formed trialkylphosphite ester may be isolated if the HBr byproduct is scavenged by a base. In the presence of HBr, a series of acid-base and SN2 reactions occur, along with the transient formation of carbocation intermediates. Rearrangement (pink arrows) of the carbocations leads to isomeric products.
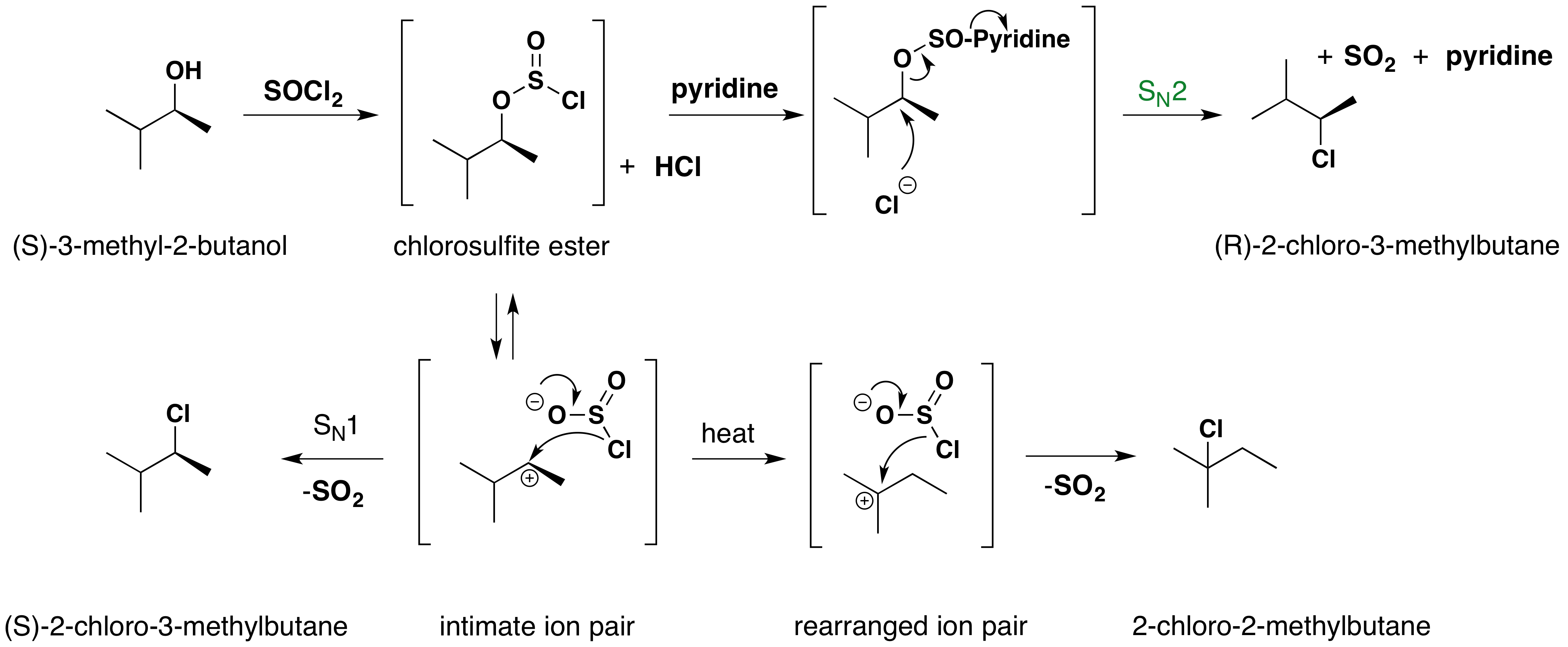
The reaction of thionyl chloride with chiral 2º-alcohols has been observed to proceed with either inversion or retention. In the presence of a base such as pyridine, the intermediate chlorosulfite ester reacts to form a "pyridinium" salt, which undergoes a relatively clean SN2 reaction to produce the inverted chloride. In ether and similar solvents, the chlorosulfite reacts with retention of configuration, presumably by way of a tight or intimate ion pair. This is classified as an SNi reaction (nucleophilic substitution internal). The carbocation partner in the ion pair may also rearrange. These reactions are illustrated by the following equations. An alternative explanation for the retention of configuration, involving an initial solvent molecule displacement of the chlorosulfite group (as SO2 and chloride anion), followed by chloride ion displacement of the solvent moiety, has been suggested. In this case, two inversions lead to retention.
Another characteristic of thionyl chloride reactions is their tendency to yield allylic rearrangement products with allylic alcohols. This fact is demonstrated by the following equations. Reactions of this type have been classified as SNi', where the prime mark indicates an allylic characteristic to the internal substitution. They may also be considered retro-ene reactions, a special class of pericyclic reactions.

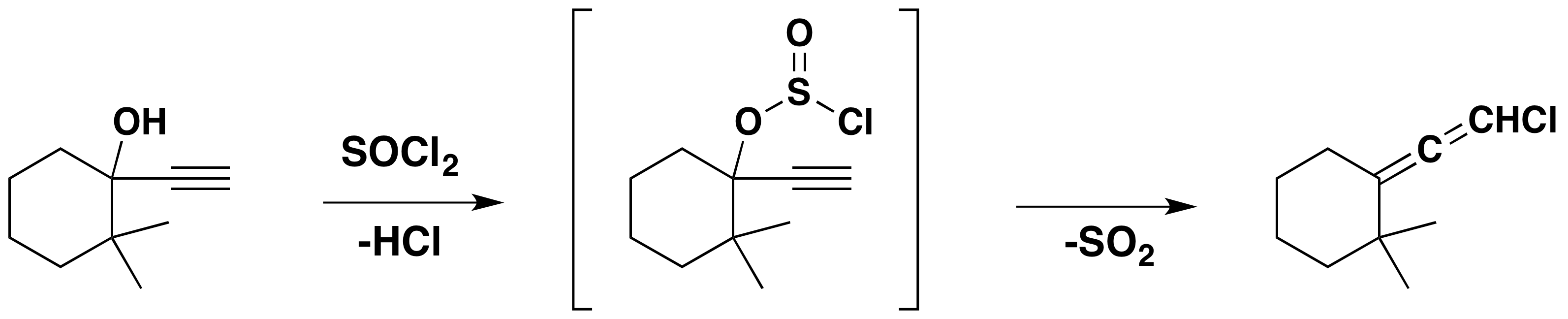
A similar substitutive rearrangement also occurs with propargyl alcohols, as shown below.
Hypervalent Phosphorous Reagents
The ability of phosphorous to assume many different valencies or oxidation states was noted elsewhere. The nucleophilicity of trialkyl phosphines allows them to bond readily to electrophiles, and the resulting phosphonium ions may then bond reversibly to other nucleophiles, especially oxygen nucleophiles. The use of phosphorous ylides in the Wittig reaction is an example of this reactivity.
The reaction of triphenylphosphine with halogens further illustrates this hypervalency. As shown in the following diagram, triphenylphosphine (yellow box on the left) reacts to form a pentavalent dihalide, which is in equilibrium with its ionic components in solution.

Chemists have used these and similar reagents to achieve the mild conversion of alcohols to alkyl halides with clean inversion of configuration. As with other OH substitution reactions, an inherently poor leaving group (hydroxide anion) is modified to provide a better leaving group, the stable compound triphenylphosphine oxide. Two such reactions are shown in the following diagram. Using this approach, even sluggish alcohols that are prone to rearrangement (e.g. neopentyl alcohol) are converted to their corresponding halides.
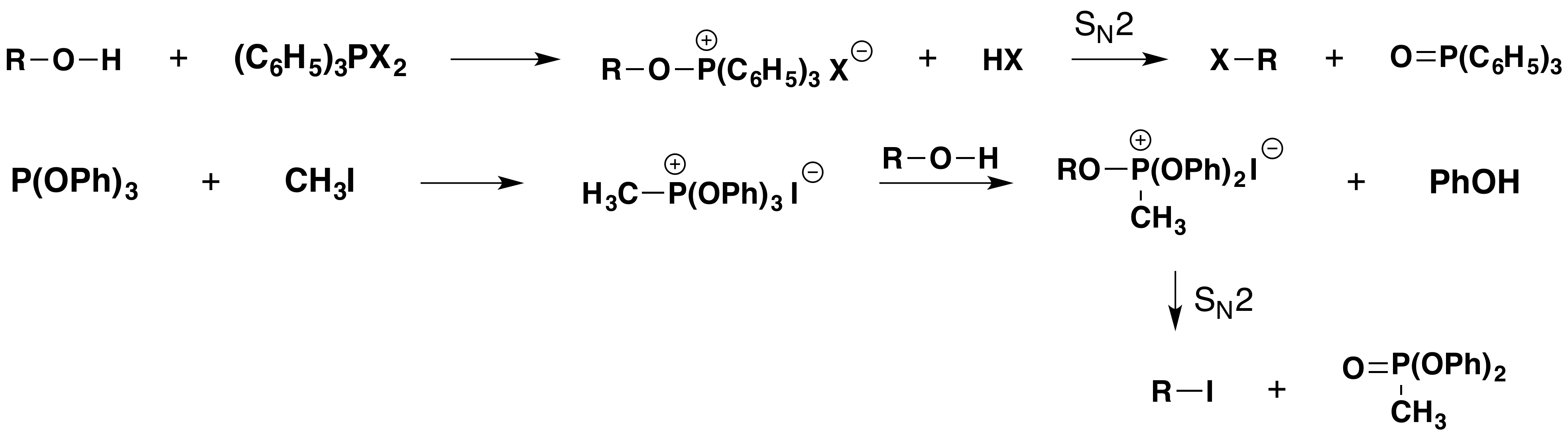
The instability of vicinal diiodides relative to their double bond analogs is the driving force for a novel transformation of vic-glycols to their corresponding alkenes. An example is displayed below.
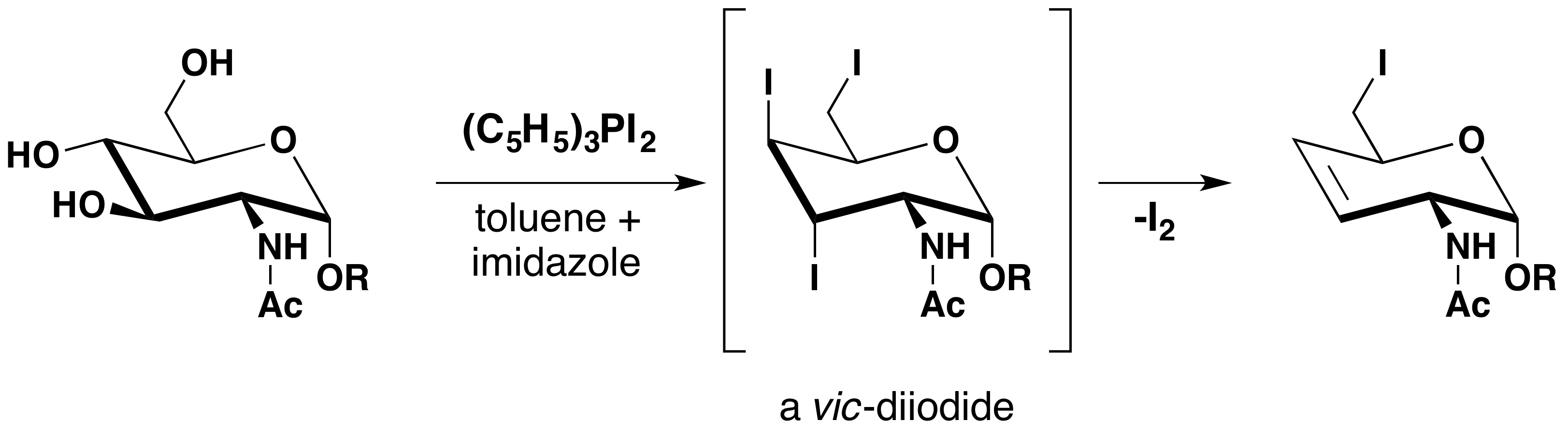
The allylic rearrangement observed in thionyl chloride is similarly avoided by using triphenylphosphine dichloride, or alternatively, by a two-step procedure via a sulfonate ester. This is displayed below.

The Mitsunobu Reaction
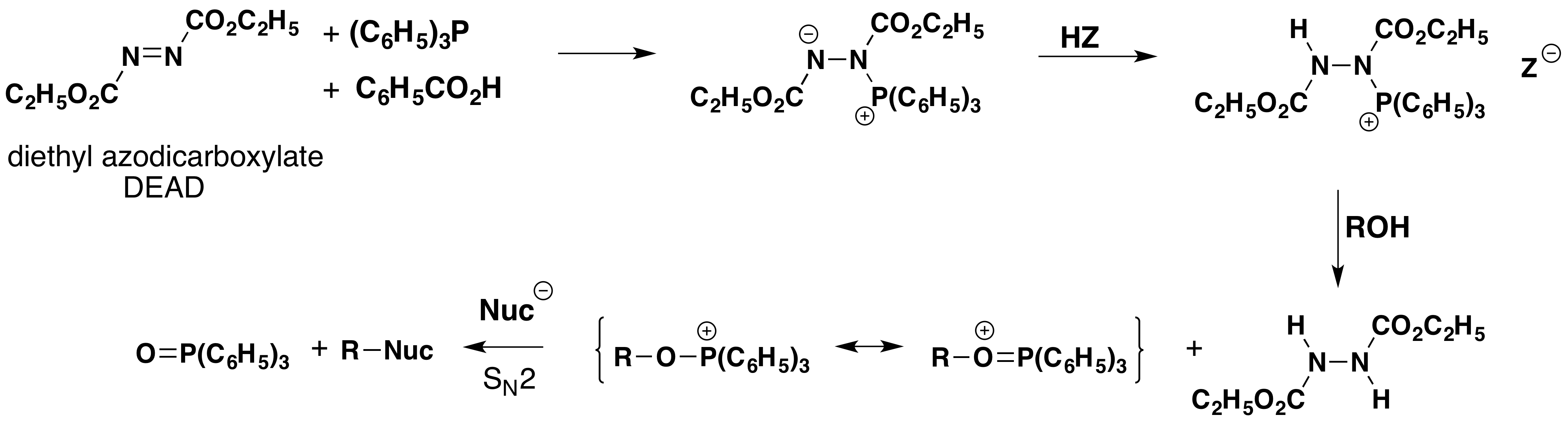
The Japanese chemist, O.Mitsunobu, devised a general and exceptionally versatile variant of hypervalent phosphorous chemistry that has been applied to a wide selection of alcohols. This method, which now carries his name, uses a reagent mixture consisting of triphenylphosphine, diethyl azodicarboxylate (DEAD) and a moderate to strong acid. The steps leading to hydroxyl substitution are outlined in the following diagram. It should be noted that the nucleophile involved in the final SN2 substitution may be the conjugate base of the acid component or a separate species.
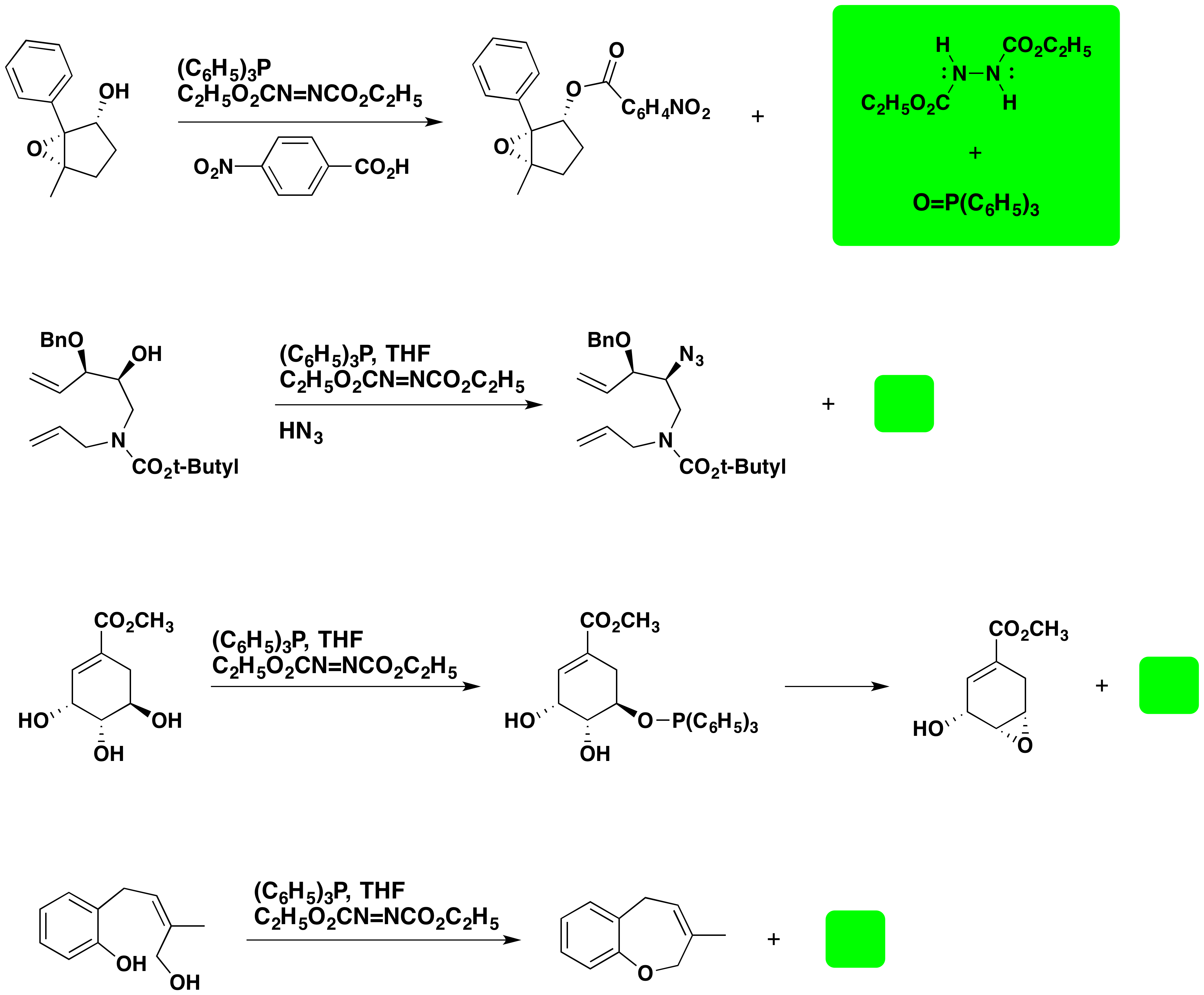
A common use of the Mitsunobu reaction is to invert the configuration of a 2º-alcohol. This application usually employs benzoic acid or a benzoate salt, and the resulting configurationally inverted ester is then hydrolyzed to the epimeric alcohol. An example of this procedure is displayed below.

In addition to achieving configurational inversion of carbinol sites, the Mitsunobu reaction has also been used to introduce the azide precursor of amines and for the intramolecular preparation of cyclic ethers. Examples are shown below.
Contributors
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry