
Appendix
- Page ID
- 59710

This rather detailed procedure is provided to the students in our Instrumental Analysis laboratory class via a website. We provide separate procedures for the experiment and operating instructions for the instruments. As noted in the main text, the students could be given far more control over the experimental design, while maintaining a robust analytical procedure.
Background on ASV
The electrochemical cell in our ASV uses 3 electrodes: a glassy carbon working electrode (WE), a silver/silver chloride reference electrode (RE), and a platinum wire counter electrode (CE). The WE is plated with a thin film of mercury immediately prior to a set of analyses (several hours of analysis) requiring a (reusable) plating solution. The analyte ions are deposited into the Hg film by applying a negative voltage to the WE during the deposition step (M2+ + 2e- = M). The potential at the WE is then scanned toward more positive potential during the stripping step. Once the oxidation potential of the metal of interest is reached, the metal is oxidized and “stripped” from the WE back into solution. The potential at which oxidation occurs is unique to each metal, enabling identification of the metal (i.e., qualitative information). The current produced from each metal (peak area or peak height) is directly proportional to the ion’s charge times the concentration in the sample solution (i.e., quantitative information). Variable deposition times enable the ppb-level measurement of many metals in aqueous solution.
Instrumentation/Equipment
Cogent/Modern Water PDV6000plus
Ultrasonic bath
Automatic micropipettes (2 – 20 μL, 20 – 200 μL, and 1000 μL)
Chemicals
- CLAC electrolyte (acetic acid/acetate buffer with sodium chloride)
- Reference electrode fill solution (saturated KCl)
- Electrode conditioning solution (proprietary, but contains 0.1 M NaOH)
- Hg film plating solution (100 ppm Hg(NO3)2 in CLAC)
- 20 ppm Cd and Pb standards (we also prepare a mixed 10 ppm Pb + Cd standard by mixing equal volumes of the two standards)
- Deionized water (18 MΩ)
- Concentrated nitric acid (reagent or ICP grade)
(Note: items 1 to 5 are available for purchase from Modern Water, Inc.)
Sample Preparation
Ultrasonic extraction of the samples is carried out in a nominally 1.5 M HNO3 solution, according to the following procedure: 1) two cigarettes are peeled and the tobacco weighed; 2) tobacco leaves are placed into a clean 50-mL beaker with 20 mL of nitric acid (1:10 dilution of concentrated HNO3); 3) the beaker is placed in an ultrasonic bath (without heating) for 15 minutes; 4) the aqueous sample is gravity filtered through a funnel with filter paper and transferred into a clean analysis cup. For the analysis, a 1.00 mL aliquot of the sample extract is combined with 20.00 mL of the CLAC electrolyte solution.
Analysis Protocol
The analysis is conducted according to the instrument manufacturer's specified procedures. Briefly, the working electrode is polished and cleaned and then inserted into the electrochemical cell and connected to the potentiostat. (The electrodes, receptacles and connections are all color-coded.) The other two electrodes are rinsed with DI water and inserted and connected. The WE is plated with the Hg solution at -1300 mV (all potentials vs. Ag/AgCl) for 5 minutes.
The VAS software uses a nested file structure that holds the instrument configuration file (including the potential vs. time waveform) and separate sample files in a project folder; each sample file contains the blank, sample, and multiple standard addition voltammograms, and a results summary (if the analysis is complete). All voltammograms within a sample file are recorded with the same waveform/configuration; we use a 600 second deposition step at -1300 mV and a linear scan from -850 to -50 mV for the stripping step.
During the analysis students record a blank voltammogram using 20.00 mL of CLAC electrolyte, add the sample aliquot through the hole in the cell and record this as the sample, and complete the analysis by adding spikes of the applicable standard, running a standard addition voltammogram each time. The software can display one voltammogram or a stacked plot and running the analysis causes the standard addition plot to be displayed with derived concentration values. Students then extract the data (current peak area and height) by double-clicking on any identified peak in an individual voltammogram and analyze it in a separate Excel file.
Hazards
Nitric acid is a strong oxidizing acid that can cause severe burns. All of the sample handling during the extraction must be performed in a fume hood and nitrile gloves worn at all times. Cadmium is carcinogenic to humans. Lead is a poisonous metal that can target the nervous system. Waste from the analysis must be placed in the labeled container for disposal. The mercury plating solution is reused five times and then placed in the mercury waste container. Cigarettes may become contaminated with toxic chemicals and thus should only be used for laboratory experiments.
PreLab Calculation
Students must construct a spreadsheet to analyze the following set of representative data in advance of the lab, which the TA will review and correct before allowing the students to begin the lab experiment. The requested analysis (including errors) is described below.
Sample Data
Table SI 1 - “Challenge unknown” sample data
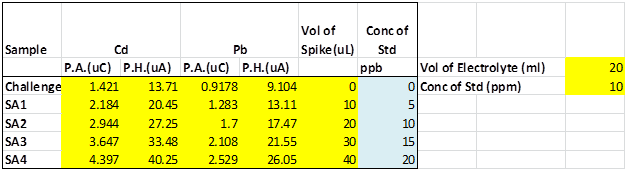
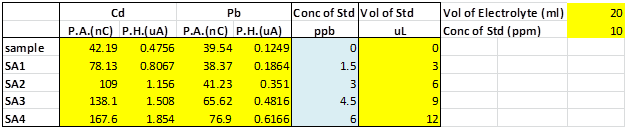
Table SI 2 - “Cigarette test sample” sample data
Operating Instructions for Cogent/Modern Water PDV6000plus and VAS software
Important Note 1 - DO NOT turn on the PDV6000plus WITHOUT placing the electrodes in a sample cup containing a suitable electrolyte as described below. The resulting voltages can damage the Working Electrode.
Important Note 2 - DO NOT invert the electrode holder assembly because liquid can destroy the stirrer motor. (It is tempting to turn it over when you separate it from the rest of the cell.) Do not fill the analytical cups with more than 25 mL of liquid since that can cause liquid to splash up into the motor. Do not leave liquid in the cell as liquid will evaporate and condense in the motor).

Figure 1 PDV6000Plus and Laptop computer with VAS software
Special notes for handling the electrodes
- Electrochemistry is sensitive to contamination from many sources; nitrile gloves must be worn at all times.
- Excepting the instructions to wipe the WE surface with filter paper, rinse it with DI water, and to polish it with the cleaning pad; do not touch the WE surface to anything, especially your fingers (even with gloves). Any physical contact can cause contamination and/or scratches that can damage the WE surface and affect the quality of the results.
1. Prepare the Working Electrode (WE)
Do this before you turn on the instrument
a) Remove the working electrode from the plastic storage vial (the electrodes are wrapped in a piece of tissue) – the working electrode is the one with the blue cap. Gently remove the blue cap; being careful not to touch the electrode surface.
b) Using the polishing kit supplied put a few drops of the polishing solution on the pad inside the cap of the kit and gently polish the electrode surface using a circular or figure-eight motion for 20 seconds.
c) Rinse the WE with deionized water and gently wipe the electrode surface on a clean piece of filter paper.
d) Visually confirm that the black electrode surface is clean and shiny with no visible scratches.
e) Dip the electrode into Electrode Conditioning Solution A for 20 seconds then rinse with deionized water and install the electrode as described in the next step.
2. Install the electrodes and prepare the cell for plating
a) Pry the black plastic cap off the electrode holder on the electrochemical cell (the cell is the cylindrical apparatus in the metal mount on the side of the yellow case). Insert the WE through the hole marked with the blue dot, being careful to not scrape the electrode surface. Install the blue wire on the metal connector on the top of the WE.

Figure 2 Color codes for different electrodes (top view)
b) Take the other two electrodes out of the storage container and carefully remove the protective caps (red and white) from the other two electrodes. Again, do not touch the electrode surfaces, since oils from your fingers will seriously contaminate the electrodes resulting in poor performance.
c) Rinse the CE (with the red top) with DI water, install it in the appropriate receptacle, and install the red connector.
d) Ensure that the reference electrode (white top) is full of electrolyte without bubbles and that the silver wire is an even brown or black color. If not, notify your TA and ask for assistance. Install the RE in the final receptacle and install the white connector.
e) Replace the black plastic cap on the top of the cell.
f) Pour the whole bottle (25 mL) of the Mercury Plating Solution into the labeled analysis cup.
g) Remove the electrode holder from the analytical cup holder. (The cell comes apart with a gentle twisting motion, leaving the clear plastic and bottom part separated from the part with the electrodes and cable.) Being careful not to pull the cable too much or to set the electrodes down on the table, place the cup with the Mercury Plating Solution in the bottom of the analytical cup holder, and reassemble the cell. In our experience, it is easier to insert the cups by holding them on only one side with a thumb and forefinger.
3. Turn on the instrument
a) Verify that the cell cable is plugged into the PDV6000Plus, the grey cable is connected to the USB to serial connector and thence to the computer, and that the power supply is plugged into the center top of the PDV6000plus.

Figure 3 Front panel of the instrument (On button shown)
b) If they are not already on, start the VAS software (under the Start button in the VAS folder) and turn on the PDV6000plus (push the On button on the device). From the Instrument menu in VAS, select Connected Instrument…. Then select PDV6000plus from the Instrument Type group. Switch to the Connections tab and click the Test Communications button to confirm communications with the analyzer and OK if the connection is verified. If there are problems, ask the TA for help.
c) Create a new Project (select New under the Project menu item in VAS) and use some reasonable name that identifies your group (all experiments by your group will end up in this project but all other student groups will have similar project folders).
d) Open the “CH427PbCdexpt” project and copy the “CH427PbCdexpt” configuration file item from near the top of the list. Close the CH427PbCdexpt project. Paste the CH427PbCdexpt configuration item into your project and double-click on it to examine the voltage waveform that is actually being used. You can print this to a file using the pdfCreator printer. Click Set Active to activate the configuration and then close the configuration window.
4. Plate the working electrode
a) From the Instrument menu, select Condition Electrode
b) Plate a thin mercury film on the electrode using a plate potential of -1300 mV for 300 seconds and a rest potential of -100 mV (you may need to enter these numbers in the dialog box).
c) Return the plating solution to the bottle (they can be used 5 times) and make a mark on the side of the bottle next to the word “used”. (If there are already 5 marks, don’t use the plating solution again. Alert the TA and they will get a new plating solution bottle for you.)
d) Rinse the cell by placing the appropriately labeled cup with ~25 mL of deionized water in the analyzer and clicking the rinse button (the little blue cup icon on the toolbar) or by choosing Rinse Cell from the Instrument menu. Rinse for 30 seconds the first time.
e) Replace the deionized water (you can reuse the same cup multiple times, just rinse it well with deionized water between uses) in the cup and do a 5 second rinse. Again replace the DI and do another 5 second rinse. We will call this whole procedure (3 rinses, one 30 sec and two 5 sec) a “rinse” and unless specifically told not to, you should always rinse before each new sample to avoid contamination by the plating solution or carry-over between samples.
5. Analysis procedures
The VAS software offers the option of Standard Comparison or Standard Addition quantitation modes. We will use only Standard Additions, since the Standard Comparison is more of a screening method.
5.1 Challenge “Unknowns”
1. Pipet 20.00 mL of CLAC electrolyte into a sample cup and place the cup into the cell. Assemble the cell. This will be used as the blank for the experiment and the "unknown" will be prepared by adding an appropriate standard to the CLAC. {Note: Three challenge "unknown" samples will be tested, but you will make all of them as you go along, by using a micropipette to add standards to the CLAC (after collecting a blank voltammogram) through the small hole in the side of the electrochemical cell.}
Table 1. Challenge unknowns
|
Name |
Description |
CLAC Electrolyte |
Standard solution |
|
SA1 |
10 ppb Cd sample |
20.00 mL |
20 ppm Cd |
|
SA2 |
10 ppb Pb sample |
20.00 mL |
20 ppm Pb |
|
SA3 |
10 ppb Cd/Pb sample |
20.00 mL |
10ppm Cd+Pb |
PreLab Calculation 1: Calculate the appropriate amount of standard solution to be added to the 20.00 mL of CLAC and verify your calculations with your TA.
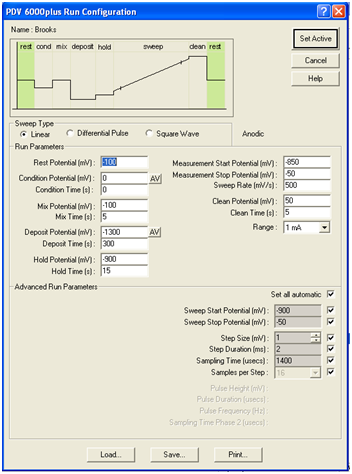
Figure 4. Run Configuration
2. Create a new sample file (select New from the Sample menu) and give it a name (e.g., SA1_Group 1_Cd). From the Instrument menu, select Active Run Configuration, and verify that CH427PbCdexpt is the active run configuration and that the Deposit Time is 300 s as shown in Fig. 4. Click Set Active and close the configuration.
3. Verify that your sample file name is still highlighted, then from Instrument menu select Initiate Run.
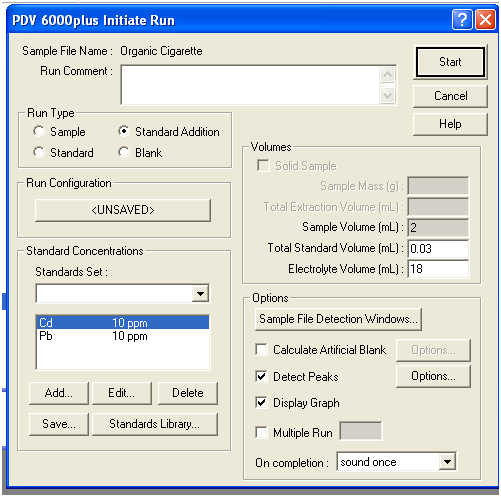
Figure 5. Initiate Run dialog box
4. In the Initiate Run dialog window, click the radio button for Blank under Run type, and the Display Graph box under Options. The other boxes may or may not have the information shown above, it is not needed for a blank.
5. Click Start to initiate the run. The voltammogram display will open and you will eventually (after about 5 minute) see the blank voltammogram. If you see any peaks in the voltammogram, you should alert the TA (this is an indication of contamination).
6. Add the appropriate amount of the correct standard (according to your calculations in response to Table 1) to produce your first challenge unknown sample. This volume should be some number of microliters and will be added to the CLAC using a micropipette inserted through the hole in the side of the cell. Be careful that the whole sample ends up in the sample cup.
7. From the Instrument menu select Initiate Run, set Run Type as Sample, and make sure the Detect Peaks and Display Graph boxes are checked.
8. Enter 20mL for Sample Volume and 0.1mL for the Electrolyte Volume (20mL plus the volume you added in step 6 is the actual amount of your sample, but the program requires a non-zero electrolyte volume). Then click Start to initiate the run. In this case, the voltammogram should show a clear peak or peaks for the standard that you added to create your challenge unknown.
PreLab Calculation 2: The spike concentration used for a standard addition should increase the analyte’s concentration by about one half of the estimated starting concentration (e.g. if you used 20ppb for both metals, you would add 20uL of 10ppm standard to 20mL of sample in the cup, then the concentration in the sample cup would increase to 30ppb after the spike.) This results in a doubling of the original signal in two spikes and an exceedance by the third, generally producing linear SA plots without saturation. Calculate the appropriate spike volumes for your challenge unknowns.
9. Add the spike volume you calculated, again using the micropipette to reach through the hole in the cell.
10. Select Initiate Run from the Instrument menu, set Run Type to Standard Addition, and make sure the Detect Peaks and Display Graph boxes are checked. Enter the total spike volume* in the Total Standard Volume box, and verify that the concentrations of the standards are correct in the drop-down box labeled Standards Set. Click Start to initiate the run. *(If you are adding 10 uL spikes, you would enter 0.02 mL for the second spike and 0.03 mL for the third and so on.)
11. Repeat steps 9 and 10 to collect a total of three or four standard additions. You can close the voltammogram display by clicking the lower red "X" in the upper right corner of the program.
12. Double-click the sample file name in VAS to show a graph of all voltammograms overlaid. Verify that the detection windows cover the peaks for each of the metals of interest (this depends on which challenge standard you are running, it can be either one metal or both metal, in which case Cd is to the left - more negative potential). If not, you can click on the bar near the top of the display to move or expand the windows. Verify that the baselines of the peaks are well-represented by the line(s) across the bottom (you can modify them by clicking on either end of the baseline and moving them).
13. From the Analysis menu select Calculate Result (if a peak is not highlighted, select Detect Peak from the Analysis menu first). The Standard Addition plot is shown and the concentration is given, based on the dilution information provided (in this case, no dilution – the concentration that you targeted). Record this value in your notebook.
14. Manually extract the raw data by double-clicking on the current peak(s) in each voltammogram; record the Peak Area and Peak Height in your notebook or directly enter them into Excel. Perform the Analysis of the Standard Addition as described below and verify that the results are comparable to those produced by VAS.
15. Repeat the whole procedure for the next Challenge Unknown: Dispose of the sample (in the proper waste container) and do a Rinse procedure. Start with a clean cup full of CLAC which serves as the blank and the diluent for the sample. Create a new sample file (reflecting the second challenge unknown) within the same project and repeat the procedure above from step 3 on, using the second "recipe" you calculated from the information in Table 1. After the analysis of the second challenge unknown, repeat the whole procedure for the final challenge unknown and then conduct a rinse before proceeding to the cigarette sample.
5.2 Cigarette sample analysis
1. Create a new sample file (select New from the Sample menu and give it a logical name).
2. Again use 20.00 mL of CLAC electrolyte as the blank and place the cup in the cell; verify that the correct sample file name is highlighted in the VAS project window, and click Initiate Run from Instrument menu; in the dialog window, check Blank for the Run Type and make sure the Display Graph box is checked.
3. Using the 1000 uL micropipette, add 1.0 mL of the cigarette sample extract into the sample cup through the hole, making sure to deliver all the liquid into the sample cup.
4. From the Instrument menu click Initiate Run; in the dialog window, check Sample for the Run Type and make sure the Detect Peak and Display Graph boxes are checked; Enter the Sample Volume 1 mL and Electrolyte Volume 20 mL; click Start to initiate the sample analysis.
5a. Choosing a spike volume requires some judgment, so it may be prudent to consult with the TA. Based on the peak sizes for the two metals (or in some cases, only one metal) and the results for the challenge unknowns above, estimate the concentration of lead and cadmium in your sample. You may need to use the zoom tool (magnifying lens icon) to see the smaller peak if there is a big difference between the two. The spike concentration for the SA method should again be about one-half of the estimated concentration (e.g. if you estimated 20 ppb for both metals, you could add 20 uL of the 10 ppm standard to the 20.00 mL of sample in the cup, raising the concentration from 20 ppb to 30 ppb). If you are uncertain about the amount of standard to use to spike the sample, always choose a smaller volume - once you add a spike, you can’t remove it.
5b. If both peaks are absent from the sample (this happens sometimes with real world samples) add only two 10 ppb spikes, allowing you establish the method limit of detection. In this case you would report the metals as ND (non-detectable at given LoD) and report your derived LoD next to your ND result.
6. Add the spike volumes you estimated to the sample cup; click Initiate Run from the Instrument menu and set Run Type to Standard Addition; make sure the Detect Peaks and Display Graph boxes are checked. Enter the total spike volume in the Standard Volume box, and verify the concentration of the standard is correct in the drop-down box. Click Start to initiate the run.
7. Repeat the spiking procedure as above for three or four standard additions and process the data as you did for the challenge unknown, except that the concentration you obtain in this case is that of the tobacco extract. This will then be scaled up to the mass in each cigarette by multiplying the concentration by ten. {Concentration in ppb, taken as ng/mL, times volume of extract (20 mL) gives mass in ng, divided by two cigarettes.}
Error Analysis
The VAS software provides concentrations for each analyte from its built-in Standard Addition procedure, but does not provide an estimate of the uncertainty of those results or provide the students insight about the calculation. Students are thus required to extract the peak height and area information from the voltammograms and perform a regression analysis in Excel to obtain the results with uncertainty (standard errors).
We assume a linear relationship between signal R (peak height or area) and added analyte concentration C*
\[R= mC^*+b\tag{A-1}\]
and a total spike volume Vs
\[C^*= \dfrac{C_s V_s}{V_x+V_s}\tag{A-2}\]
where Cs is the concentration of the standard used to spike the unknown sample and Vx is the volume of the unknown. The y-intercept b in Eq. A-1 is the signal produced by the original (unknown) concentration of the analyte and the slope m represents the Response Ratio – the increase in signal per unit increase in concentration of the analyte. The absolute value of the x-intercept (where R = 0 in Eq. A-1) is the desired concentration Cx of the unknown – the amount of analyte that would have to be removed from the system to make the signal go to zero.
In order to obtain the statistics for the fit (used to establish the uncertainty) along with the concentration, the linear SA plot is constructed in Excel and the regression line is obtained: 1) instrument response (peak height or area) is plotted against the concentration increase of the analyte (obtained from Eq A-2) and then 2) the best fit line is obtained using Excel's Regression Analysis in the Data Analysis Toolpak. The x-intercept could be obtained from the plot by graphical extrapolation but here we obtain the best fit value for m and b and then divide the intercept by the slope to obtain the desired concentration.
\[C_x = \dfrac{|b|}{m} \tag{A-3}\]
To determine the uncertainty in this concentration, the standard propagation of error rule for multiplication and division (“relative variances add”) is used
\[\left(\dfrac{S_C}{C_x} \right)^2 = \left(\dfrac{S_b}{b}\right)^2+ \left(\dfrac{S_m}{m}\right)^2 \tag{A-4}\]
where Sc is the desired standard error for the unknown concentration, Sm is the Standard Error of the slope (X variable 1 in the Data Analysis Regression) and Sb is the Standard Error of the Intercept.