
Gas Chromatographic Separation Methods
- Page ID
- 71837

A gas chromatograph typically has a carrier gas (it is common to refer to the mobile phase in gas chromatography as the carrier gas since the mobile phase has no influence at all over the separation – its only purpose is to carry the solutes through the column), heated injection port, column in a heated oven, and detector.
The carrier gas must be inert and helium or nitrogen is used for most gas chromatographic applications. Sample injection is most commonly accomplished through the use of a small-volume syringe (1 μL is a typical injection volume). The liquid sample is injected into a hot zone that is sealed with a polymer septum. The syringe pierces the septum, and on injection the liquid sample is flash-volatilized in the heated injection port and flushed into the column by the carrier gas. With fused silica capillary columns, 1 μL of liquid sample will overload the column so the injector is designed with a splitter. Adjusting the flow of the splitter gas allows you to calibrate the split ratio. Ratios of 50:1 to 100:1 (only the one part is injected) are common. Most injection ports of this type have glass liners. Different glass liners are inserted under the septum depending on whether you are doing split or splitless injections.
Another gas chromatographic injection technique that is used in certain applications involves thermal desorption from a polymer trap. The technique is illustrated in Figure 70 for the analysis of organic chemicals in air. Assume that we have a small piece of tubing filled with a polymer. The polymer is a material that organic chemicals will adsorb to. Water in the air will pass through unretained. A polymer that is used quite frequently for this application is Tenax. Tenax is a p-polyphenylene oxide polymer that is stable at very high temperatures. In the adsorption step, we use a pump to draw some large volume of air (this might be as high as 15 liters of air) through the Tenax trap, which is held at room temperature. The organic chemicals from the air will adsorb onto the polymer.
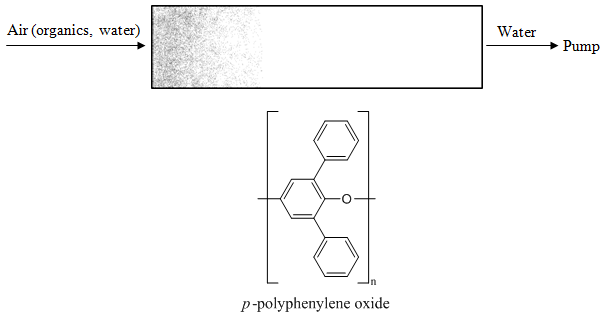
The organic chemicals can be desorbed from the Tenax trap by heating the gas under a flow of helium. The illustration in Figure 71 shows how the Tenax trap is desorbed in a backflush mode (the end that had the higher concentrations of organic chemicals is put closer to the gas chromatograph). Switching valves comparable to liquid chromatographic injection valves make it easy to redirect the flow of gases through these Tenax traps between the adsorption and backflushing orientation.
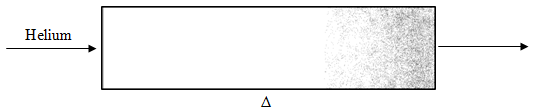
Desorption might typically be done at a temperature of 225oC over a period of five minutes. One thing to realize is that we would never be able to tolerate a five-minute injection of a sample, since this would mean that all of the peaks might be as long as five minutes. It is essential to maintain a “plug” injection, which is achieved by thermal focussing. Thermal focussing is accomplished by cooling a region in the injector (or in older gas chromatographs, the entire oven) to a temperature of –50oC. This cooling is accomplished using either liquid carbon dioxide or liquid nitrogen. At –50oC, the organic compounds that desorb from the Tenax trap freeze in a small band at the head of the column. Rapid heating of this frozen band injects the sample at a comparable rate to a normal syringe injection.
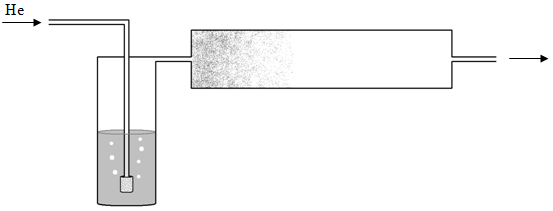
Sorbent traps are frequently used for the analysis of trace levels of volatile organic chemicals in water as well. In this case, the method is known as a purge and trap technique and is shown in Figure 72. A sample of water is taken (usually 5 mL – too large to ever inject into a gas chromatograph) and purged with helium gas. After exiting the water, the helium gas flows through a Tenax trap. The helium gas bubbling through the water displaces the dissolved volatile organic chemicals, which adsorb onto the Tenax trap. The Tenax trap is desorbed as described above for the analysis of volatile organic chemicals in air.
Information on a variety of commercially available gas chromatographic stationary phases is provided in Table 3. The distinction between different stationary phases is based on a comparison of their polarity. Five different compounds are typically used to represent different types of functional groups. Individual indices for these five compounds are measured, and a composite value (P) is determined as well. These polarity indices are referred to as Rohrschneider constants. The higher the number the more polar the phase. In reality, a sampling of four or five stationary phases would be good enough to span the range of polarities that is needed for gas chromatographic separations.
Table 3. Rohrschneider constants for gas chromatographic liquid phases
| Liquid phase | Max T oC |
X' | Y' | Z' | U' | S' | P |
|---|---|---|---|---|---|---|---|
| Squalane | 100 | 0 | 0 | 0 | 0 | 0 | 0 |
| Apiezon L | 250 | 32 | 22 | 15 | 32 | 42 | 29 |
| SE-30 | 300 | 15 | 53 | 44 | 64 | 41 | 43 |
| OV-1, methyl gum | 350 | 16 | 55 | 44 | 65 | 42 | 44 |
| OV-3, 10% phenyl | 350 | 44 | 86 | 81 | 124 | 88 | 85 |
| OV-7, 20% phenyl | 350 | 69 | 113 | 111 | 171 | 128 | 118 |
| Dioctyl sebacate | 125 | 72 | 168 | 108 | 180 | 123 | 130 |
| Dilauryl phthalate | - | 79 | 158 | 120 | 192 | 158 | 141 |
| Dinonyl phthalate | 150 | 83 | 183 | 147 | 231 | 159 | 161 |
| OV-17, 50% phenyl | 375 | 119 | 158 | 162 | 243 | 202 | 177 |
| Versamid 930 | 150 | 109 | 313 | 144 | 211 | 209 | 197 |
| Trimer acid | 150 | 94 | 271 | 163 | 182 | 328 | 218 |
| OV-25, 75% phenyl | 350 | 178 | 204 | 208 | 305 | 280 | 235 |
| Polyphenylether | 225 | 182 | 233 | 228 | 313 | 293 | 250 |
| Triton X-305 | 200 | 262 | 467 | 314 | 488 | 430 | 392 |
| Carbowax 20M | 225 | 322 | 536 | 368 | 572 | 510 | 462 |
| Carbowax 4000 | 200 | 317 | 545 | 378 | 578 | 521 | 468 |
| Reoplex 400 | 200 | 364 | 619 | 449 | 647 | 671 | 550 |
| Carbowax 1540 | 175 | 371 | 639 | 453 | 666 | 641 | 554 |
| Diglycerol | 100 | 371 | 826 | 560 | 676 | 854 | 657 |
| EGSS-X | 200 | 484 | 710 | 585 | 831 | 778 | 678 |
| Ethylene glycol phthalate | 200 | 453 | 697 | 602 | 816 | 872 | 688 |
| Diethylene glycol succinate | 200 | 496 | 746 | 590 | 837 | 835 | 701 |
| Tetrahydroxyethylenediamine | 150 | 463 | 942 | 626 | - | 893 | 731 |
| Hexakis(2-cyanoethoxycyclohexane) | 150 | 567 | 825 | 713 | 978 | 901 | 797 |
| N,N-bis(2-cyanoethyl)formamide | 125 | 690 | 991 | 853 | 1110 | 1000 | 929 |
X' = benzene; Y' = butanol; Z' = 2-pentanone; U' = nitropropane; S' = pyridine
P = (X' + Y' + Z' + U' + S')/5
If we consider what governs retention order in gas chromatography, there are two important parameters. One is the volatility of the compound, with the observation that more volatile compounds (those with a higher vapor pressure or lower boiling point) elute first. The other is the attractive forces between the compound and the stationary phase. If we consider the homologous series of alcohols listed in Table 4, it’s interesting to note that the boiling point goes up by approximately 20oC for each additional methylene group (CH2) in the chain. The increase in boiling point with each additional methylene group is a little higher for the series of alkanes. The alcohols have higher boiling points than the corresponding alkanes because they can hydrogen bond with each other.
Table 4. Boiling points for homologous series of alkanes and primary alcohols.
| Hydrocarbon | Boiling Point (oC) | Alcohol | Boiling Point (oC) |
|---|---|---|---|
| Pentane | 36 | 1-Pentanol | 137 |
| Hexane | 69 | 1-Hexanol | 157 |
| Heptane | 98 | 1-Heptanol | 176 |
| Octane | 126 | 1-Octanol | 196 |
| Nonane | 151 | 1-Nonanol | 215 |
| Decane | 174 | 1-Decanol | 231 |
If we consider a homologous series, it turns out that the boiling points are actually determined by the molar volumes of the molecules. Just as we observed with the size of molecules in steric exclusion chromatography, the molar volume of a molecule is the volume swept out by the molecule as it tumbles. Molecules with larger molar volumes have higher boiling points, provided the molecules being compared have identical intermolecular forces (you cannot compare hydrocarbons to alcohols). An interesting comparison is observed by looking at the boiling point of n-octane and iso-octane (2,2,4-trimethylpentane).
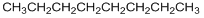
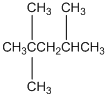
n-octane (bp = 126oC) iso-octane (bp = 98oC)
The branched iso-octane would have a smaller molar volume than the linear n-octane, and this is clearly reflected in the boiling points of the two compounds.
When predicting retention order in gas chromatography, the overriding factor is a comparison of the boiling points. The compound with the lowest boiling point elutes first. Only when two compounds have very close boiling points (within 5oC or less) does it become important to consider the polarity of the compounds and the polarity of the stationary phase. Remember that like dissolves like, so a polar stationary phase will show more retention of a polar compound. The Rohrschneider values are used to determine the polarity of the stationary phase.