
Adsorption Compared to Partition as a Separation Mechanism
- Page ID
- 70895

If we go back now and consider Tswett’s first separation, we see that he used solid starch as the stationary phase, and so solutes exhibited an adsorption to the surface of the starch. A useful thing to consider is the nature of the chemical groups on the surface of starch. Starch is a carbohydrate comprised of glucose units. The glucose functionality of starch is shown below, and we see that the surface is comprised of highly polar hydroxyl groups.
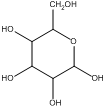
Glucose
Silica gel (SiO2) and alumina (Al2O3) were mentioned previously as two other common solid phases. If we examine the structure of silica gel, in which each silicon atom is attached to four oxygen atoms in a tetrahedral arrangement (this corresponds to two silicon atoms sharing each of the oxygen atoms), we run into a problem when you try to create a surface for this material. If we have an oxygen atom out on the edge, we would then need to attach a silicon atom, which necessitates more oxygen atoms. We end up with a dilemma in which we can never wrap all of these around on each other and only make something with the formula SiO2. Some of these groups are able to do this and some of the surface of silica gel consists of what are known as siloxane (Si–O–Si) groups. But many of the outer oxygen atoms cannot attach to another silicon atom and are actually hydroxyl or silanol (Si–OH) groups.
–Si–O–Si– –Si–OH
Siloxane units Silanol units
What we notice is that the surface of silica gel consists of very polar hydroxyl groups. It turns out that the same thing will occur for the surface of alumina.
If we consider a solute molecule (S), and have it adsorb to the surface of silica gel, we could write the following equation to represent the process of adsorption.
Si–OH + S \(\leftrightarrow\) Si–OH - - - S
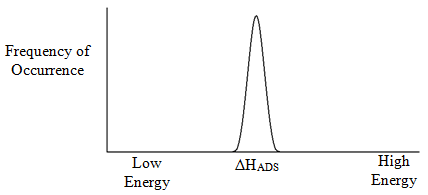
With any reaction we can talk about its enthalpy and entropy. So in this case, we could talk about the enthalpy of adsorption (ΔHADS). Suppose now we took one solute molecule and millions of surface silanols, and went through one by one and measured ΔHADS for this one solute molecule with each of these millions of surface silanols. The question is whether we would get one single ΔHADS value for each of the measurements. Hopefully it might seem intuitive to you that we would not get an identical value for each of these individual processes. Instead, it seems like what we would really get is a distribution of values. There would be one value that is most common with other less frequent values clustered around it. For some reason, some silanol sites might be a bit more active because of differences in their surrounding microenvironment, whereas others might be a bit less active. Suppose we entered all these measurements in a spreadsheet and then plotted them as a histogram with number of measurements of a particular value versus ΔHADS. But another important question involves the nature of this distribution of ΔHADS values. Would it be symmetrical as shown in Figure 15, or would it have some asymmetry?
The easiest way to think of this is to examine the nature of the silanol groups on the surface of silica gel. One thing we could ask is whether there are different types of silanols, as shown below (are there also disilanols and trisilanols?).
–Si–OH –Si–(OH)2 –Si(OH)3
It turns out that we will get some di- and trisilanols, although these are far fewer in number than the monosilanols. It should make sense that even if the distribution about a monosilanol were symmetrical, that the di- and trisilanols will have different distributions of ΔHADS. Assuming that ΔHADS is larger for the di- and trisilanols, that might lead to the plot shown in Figure 16 for each of the individual distributions and then the composite drawing for the overall distribution.
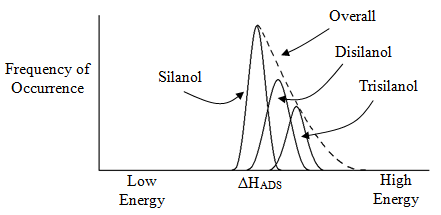
Notice the asymmetry in the plot in Figure 16 with a smaller number of highly active sites. Something to realize is that even if we had only monosilanols, there is still experimental data that shows a small proportion of highly active sites that produced an asymmetric distribution. It is important to realize that not everything in nature occurs in a symmetrical manner.
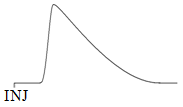
The key question for us to ask is what a chromatographic peak would look like for a solute traveling through this stationary phase. Hopefully it seems reasonable to predict that the peak for the solute would also be asymmetric. Those solute molecules adsorbed at the highly active sites would essentially get stuck on the column and take much longer to elute. The asymmetric peak observed in Figure 17 exhibits tailing. Note that tailed peaks are undesirable in chromatographic separations because they are more likely to overlap with each other and interfere with the separation. Unless the solid surface is highly deactivated, all chromatographic separations based on adsorption will exhibit peak tailing. This causes an inherent inefficiency in adsorption methods.
One last thing before we move on is to understand that strong adsorption by molecules on active solid surfaces has a profound implication in environmental chemistry. Consider a leaking, underground tank of gasoline at a service station. The soil around the tank gets contaminated by the leaking gas and it is desirable to clean it up. One way is to remove all the contaminated soil, heat it in a very high temperature oven that combusts all the hydrocarbon components of the gasoline, and recycle the soil. This is a common remediation procedure. But suppose the soil exists over a very large area (say the soil surrounding an old hazardous waste dump). This may be too large a volume of soil to use the removal and combustion method. The chemicals may have leached far away from the original site, contaminating nearby drinking wells.
Based on our knowledge of chromatography, and essentially using the ground as something similar to a chromatographic column, we could imagine pumping lots of clean water in from wells outside the contaminated area, and removing contaminated water from wells internal to the contaminated region. After some time of pumping, all the contaminants ought to migrate by a chromatographic-like process from the outside to the inside and be removed. If you do this, you eventually see that the level of contaminants in the water coming out of the inner wells drop considerably (maybe even to acceptable levels). But if you allow the system to sit for quite a while, you start to discover elevated levels of contaminants in the water again. What has happened is that some of the contaminants adsorbed quite strongly to the solid surfaces in the ground and were unavailable to the water. As the system sat, these slowly began to come off the surface into the water, raising the concentration. This kind of treatment process has generally proven ineffective as a way of completely treating such ground water contamination. It does get rid of a lot of the contaminants, but the strong adsorption requires enormous lengths of time before the levels would drop low enough.
Chromatographic methods languished under these inefficient methods for many years until Martin and Synge reported the first application of a partition separation in chromatography in 1941. Recognizing that WWII was taking place in 1941, there was a considerable need for wool clothing for soldiers from England fighting in the war. Wool is rather unique as a fabric since it retains much of its warmth even if wet. In fact, so much wool was needed that England did not have enough sheep to provide the volume of wool clothing that was necessary. Martin and Synge were interested in seeing whether it was possible to make artificial wool, and sought to examine the amino acid content of the proteins that make up wool fibers as a first step in understanding the chemical nature of wool. What they also realized, though, was that separating amino acids using adsorption chromatographic methods available at that time was going to be a difficult process. They therefore investigated whether it would be possible to take a solid particle and coat it with a liquid stationary phase, and perform the separation based on partitioning. What they found was that it was possible, and that the chromatographic efficiency was improved considerably when compared to methods based on adsorption.
The important thing to realize here is that we no longer have an adsorption process, but the dissolution of the solute into another liquid solvent. The relevant enthalpy to consider here is the enthalpy of solvation (ΔHSOLV). It turns out that ΔHSOLV of a particular solute in a particular solvent will also show a distribution of values; however, in this case, the distribution is a symmetrical one. Therefore, a chromatographic peak for a solute being separated entirely by a partitioning mechanism between the mobile and stationary phases ought to be symmetrical as well. This will greatly enhance the efficiency of the chromatography. This work was so important that it was recognized with the Nobel Prize in 1952. The only problem was that there were still other important issues with liquid chromatography such that it really could not flourish as an analytical method (we will examine these other issues later in our unit on chromatography).
One intriguing aspect of Martin and Synge’s 1941 paper is the last sentence, which predicted that it should be possible to use gas as the mobile phase (i.e., gas chromatography), and that some of the limitations that restrict the efficiency of liquid chromatography should not occur in gas chromatography. What was also interesting is that it was not until 1951, that James and Martin published the first report in which gas chromatography was described as an analysis method. Much of the delay was because of WWII and the need for scientists to devote their research to areas of immediate national concern related to the war effort.
The introduction of gas chromatography revolutionized the entire field of chemical analysis. One thing, as we will develop in the next substantial portion of this unit, is that gas chromatographic methods had certain fundamental advantages over liquid chromatographic methods when it came to column efficiency. It was possible, using gas chromatography, to separate very complex mixtures of volatile chemicals in very short periods of time. Before gas chromatography, no one even considered that it might be possible to separate as many as 50 to 100 constituents of a sample in only an hour. The other thing that prompted an explosion of interest in gas chromatography as an analysis method was the development of some highly sensitive methods of detection in the 1950s and 1960s. People were now able to sense levels of molecules that were not detectable in other, conventional solution-phase systems. Part of the problem with solution-phase analysis is the overwhelming volume of solvent that can interfere with the technique used to perform the measurement. Gas phase measurements, where the overall density of molecules is very low, do not have as much potential for interference from gases other than those being measured.
What we now need to do is develop an understanding of what created the inherent advantage in efficiency of gas chromatographic columns, and then understand what took place to improve the efficiency of liquid chromatographic columns. When we talk about the efficiency of a chromatographic column, what we really refer to is the width of the chromatographic bands of solutes as they migrate through the column.